Disease Awareness
In This Section
Epidemiology
In this section, find out about the prevalence and incidence, mortality and disease burden of idiopathic pulmonary fibrosis (IPF).
Prevalence and incidence
One of the main limitations of measuring the historical prevalence and incidence of idiopathic pulmonary fibrosis (IPF) is the absence of a uniform, consistent definition of IPF before a consensus statement was published in 2000, and then updated in 2011.1,2
IPF is responsible for 20–50% of all interstitial lung disease (ILD) cases.3
Its prevalence and incidence have appeared to be increasing over the last few decades.3
Incidence studies from 2000 onwards, in Europe and North America, estimate 3–9 cases of IPF per 100,000 per year.4
Prevalence estimates for IPF have varied from 2–29 cases per 100,000 in the general population.2

Figure 1. Key incidence information for patients with IPF (Raghu et al., 2011; Ley and Collard, 2013; Sauleda et al., 2018)
*This may be due to sex differences in historical smoking patterns rather than an inherent sex-related risk for IPF
We asked Professor Elisabeth Bendstrup why she thought that primary care physicians must maintain an index of suspicion for a rare disease such as IPF. Find out her response here.
Mortality rates in patients with idiopathic pulmonary fibrosis (IPF)
The median survival time is estimated to be two to three years from diagnosis.2
As with prevalence and incidence rates, mortality rates are higher in male patients than female and increase with age.3The most common cause of death is progressive lung disease, resulting in 60% of deaths in patients with IPF.2 Other causes include coronary artery disease, pulmonary embolism and lung cancer.2
Mortality rates appear to be increasing steadily.2 In 2014, between 28,000 and 65,000 deaths in Europe and between 13,000 and 17,000 deaths in the United States were estimated to be caused by IPF.3
Disease burden
Although idiopathic pulmonary fibrosis (IPF) is considered a rare disease, its burden is high.5
Medical costs of IPF are high
In a study of American Medicare beneficiaries, patients with IPF had a higher risk of hospitalisation (28.8 vs. 15.8%) and emergency room visits (23.9 vs. 13.1%) compared to matched control subjects.6
Total medical costs of IPF were found to be around 31,000 USD/person-year, including treatment before and after diagnosis.6
IPF heavily affects patients’ and their family’s quality of life
Patients’ and their families’ quality of life are greatly affected by:7
- poor prognosis
- disease course uncertainty
- severe symptoms.
Patients report substantial impairment on their health-related quality of life (HRQoL), especially in domains that measure physical health, activity and level of independence.8,9
Dyspnoea, cough and severity of depression particularly affect quality of life in patients with IPF, with dyspnoea the most significant contributor.8
Longitudinal data show that forced vital capacity decline is also associated with worsening HRQoL in patients.8
Despite the effect of IPF on patient quality of life, Professor Elisabeth Bendstrup recalls one of her patients who was determined to achieve his goal and managed it with the help of anti-fibrotic treatment.
Elisabeth Bendstrup answers our question ''Do you have an example of how anti-fibrotic treatment has improved patient quality of life?".
of interest
are looking at
saved
next event
Pathophysiology
In this section, learn about the pathogenesis of idiopathic pulmonary fibrosis (IPF) and the possible risk factors.
Pathogenesis
Knowledge and understanding of idiopathic pulmonary fibrosis (IPF) pathogenesis has increased substantially in the last decade and has underpinned a fundamental change in the treatment approach for patients with IPF.10
Inflammation used to be considered the main pathological driving force in IPF and corticosteroids, in addition to cytotoxic agents, were recommended at treatment.1
Recent understanding, however, suggests the pathogenesis of IPF is driven by dysfunction and maladaptive repair of the alveolar epithelium after ongoing episodes of injury (Figure 2).11,12
Activated alveolar epithelial cells release fibrogenic growth factors that promote the migration, activation and differentiation of fibroblasts and invasive myofibroblasts, which then organise into fibroblastic foci, triggering excessive collagen production that results in architectural damage and lung scarring.11
Ultimately, healthy tissue is replaced by altered extracellular matrix and alveolar architecture is destroyed, which leads to decreased lung compliance, disrupted gas exchange and finally respiratory failure and death.11,12
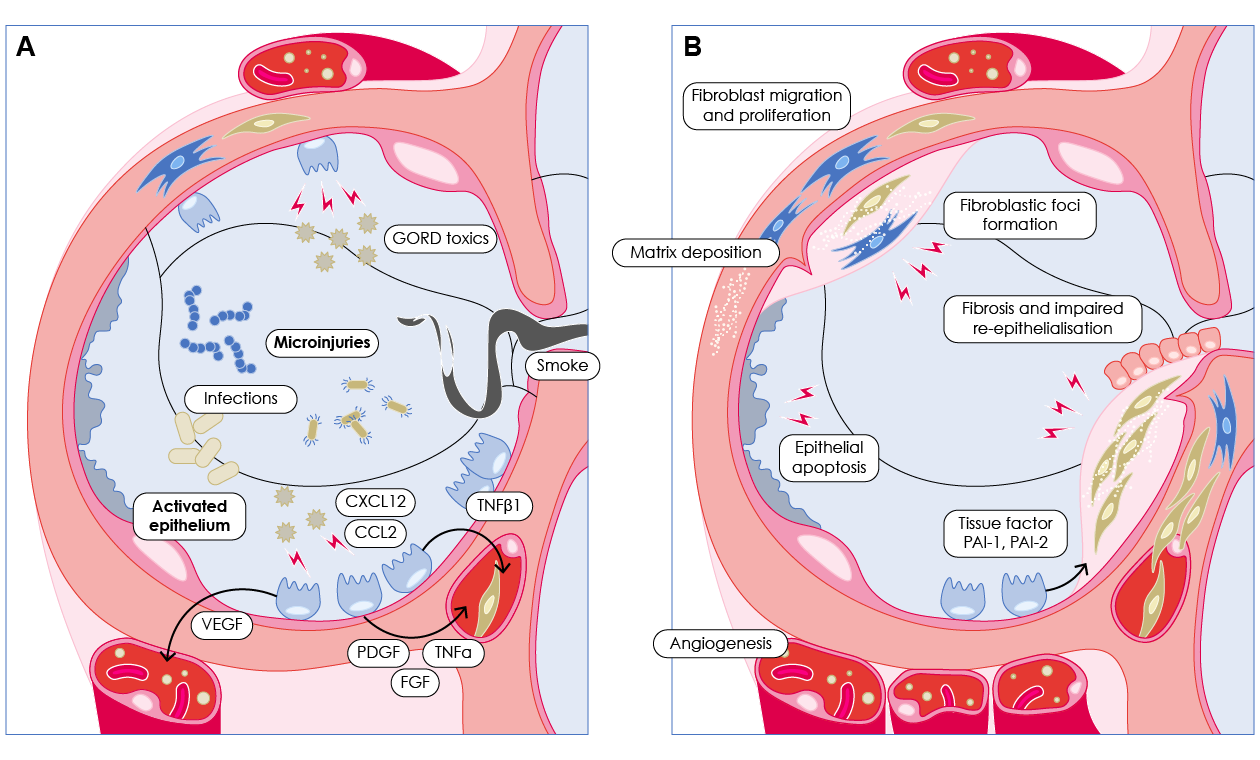
Figure 2. Schematic view of IPF pathogenesis.11 Repeated injuries over time lead to maladaptive repair process, characterised by alveolar epithelial cell apoptosis, proliferation and epithelium-mesenchymal cross-talk (a) and following fibroblasts, myofibroblasts proliferation and accumulation of extracellular matrix (b).
CCL2, chemokine C-C motif ligand 2; CXCL12, C-X-C motif chemokine 12; FGF, fibroblast growth factor; PAI-1, plasminogen activator inhibitor 1; PAI-2, plasminogen activator inhibitor 2; PDGF, platelet-derived growth factor; TGF-β1, Transforming Growth Factor-Beta 1; TNF-α, tumor necrosis factor-alpha; VEGF, vascular endothelial growth factor.
There are three pathophysiologic stages that are thought to lead to the development of pulmonary fibrosis (Figure 3).12
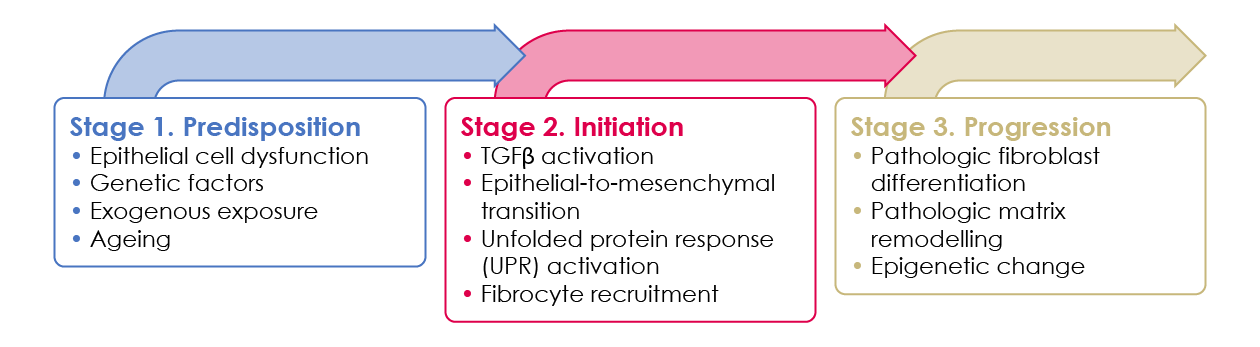
Figure 3. IPF pathogenesis can be divided into three pathophysiologic stages.12
The predisposition stage includes genetic mutations/variations that predispose individuals to develop lung fibrosis, chronic epithelial cell turnover during the lifetime of an individual (which leads to shortened telomeres) and environmental exposures. These factors may lead to epithelial cell dysfunction. Not all individuals in this stage will develop clinically relevant disease; whether they do depends on the degree and duration of exposure to these factors.
In the initiation stage, molecular mediators of epithelial cell dysfunction such as ER stress; excessive TGF-β activation; and growth factor, chemokine or Wnt secretion lead to epithelial-to-mesenchymal transition, fibrocyte recruitment and fibroblast differentiation.
These lead to the progression stage, where pathologic mesenchymal cells release abnormal types and quantities of matrix proteins that remodel and scar the lung. The pathologically remodelled matrix or epigenetic changes within fibroblasts may lead to a feed-forward loop of mesenchymal cell activation and progressive fibrosis.
Risk factors
Although the stimuli that trigger the fibrotic process in idiopathic pulmonary fibrosis (IPF) is still unknown, several factors have been associated with an increased risk of developing the disease (see Figure 4).
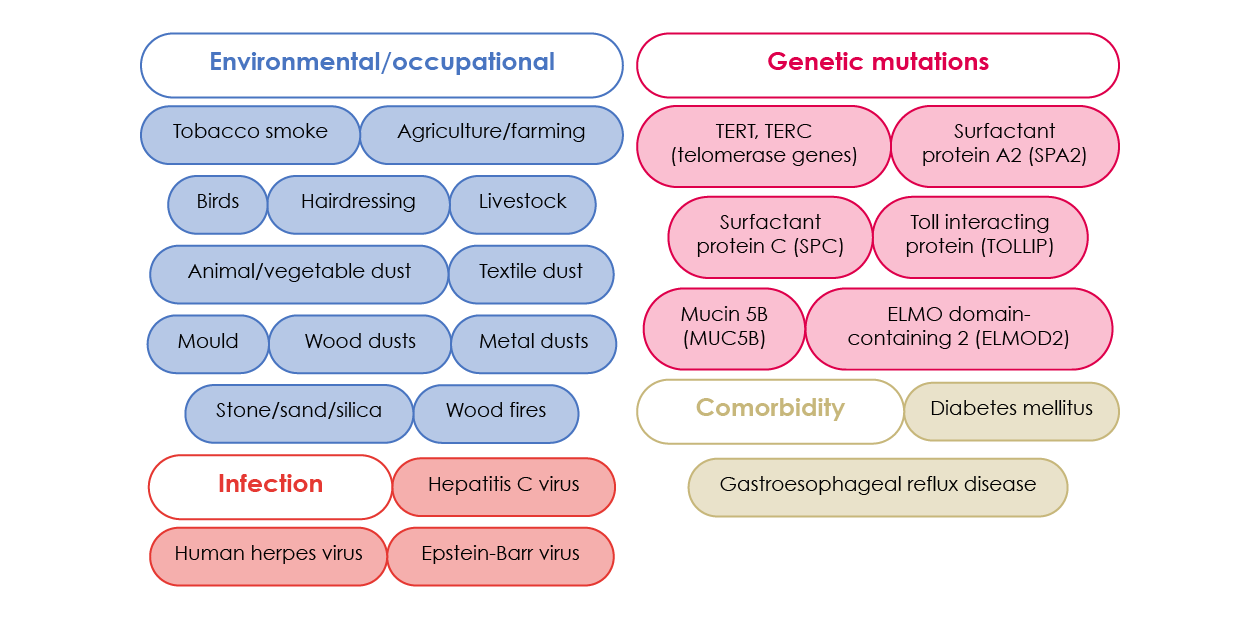
Figure 4. Proposed risk factors for IPF.2,5
Comorbidities
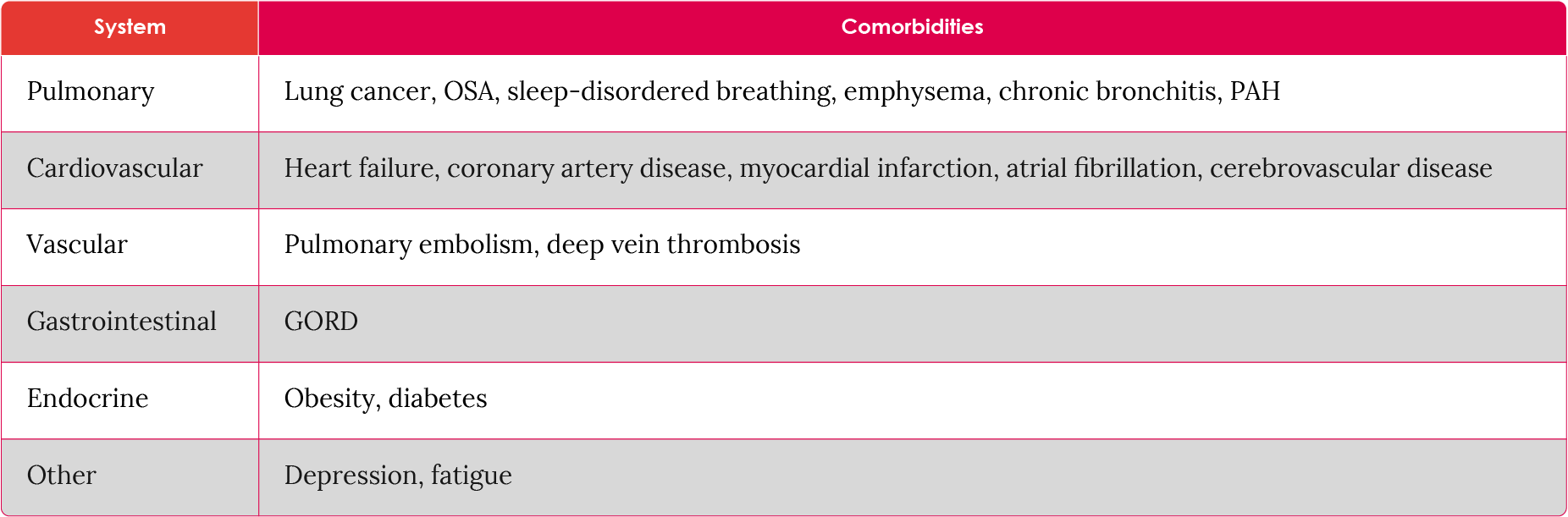
Many IPF patients have comorbidities (Table 1). For example, abnormal gastroesophageal reflux (GER/GOR) has been observed in almost 90% of patients with IPF.13
About 20–40% of IPF patients evaluated for lung transplants have pulmonary arterial hypertension (PAH), which is associated with increased mortality.14 Further, 6–44% and 15–68% of IPF patients experience mild, and moderate to severe obstructive sleep apnea (OSA) respectively15 and between 21% and 49% have depression.15
Table 1. Common comorbidities in patients with IPF.15
of interest
are looking at
saved
next event
Symptoms and referral
Patients with idiopathic pulmonary fibrosis (IPF) often present with general respiratory symptoms that can lead to an initial misdiagnosis of asthma, chronic obstructive pulmonary disease or pneumonia, among other diseases.16
Key IPF symptoms
IPF should be considered in all adult patients who have the following symptoms, in the absence of any additional symptoms that suggest a multisystem disease:17
- unexplained chronic exertional dyspnoea
- persistent cough
- bibasilar inspiratory (Velcro-like) crackles when listening to the chest
- finger clubbing
Reasons for referral delays
Delays between the onset of first symptoms and referral to a specialist centre are common with patients with IPF and can be due to:16,18
- patient-dependent factors (for example, reluctance to acknowledge symptoms that may indicate health problems and a sedentary lifestyle masking dyspnoea at exercise)
- disease-dependent factors (for example, progressive onset and slow progression of IPF allows the disease to go undetected unless exacerbations occur)
- physician-dependent factors (for example, lack of awareness of rare diseases by general practitioners and even by lung specialists)
Bibasilar inspiratory (Velcro-like) crackles
The detection of bibasilar inspiratory (Velcro-like) crackles during chest auscultation, which are strongly associated with the presence of lung fibrosis, has been proposed as a potential measure to improve early detection of IPF.16,18
Diagnosis
Diagnosing idiopathic pulmonary fibrosis (IPF) early in the disease course is important as it allows the patient to minimise any risk factors and to begin anti-fibrotic treatments that can potentially slow disease progression and prevent irreversible lung damage.16
Accurately diagnosing IPF can be challenging and often requires the collaborative expertise of a:17
- consultant respiratory physician/pulmonologist
- radiologist
- histopathologist
IPF is defined as a fibrosing interstitial lung disease (ILD) of unknown cause with the histopathologic and/or radiologic pattern of usual interstitial pneumonia (UIP).17 However, UIP is not only found in patients with IPF, but is also seen in a number of other conditions, including connective tissue diseases, chronic hypersensitivity pneumonitis and asbestosis.17
See the video 'Do you have any anecdotes showing why accurate diagnosis is important?' where Professor Kristin Highland describe a patient who was initially thought to have IPF but subsequently an underlying autoimmune cause was identified. This highlights the importance of accurate diagnosis.
The diagnosis of IPF therefore initially requires the exclusion of all known causes of usual interstitial pneumonia.17
International guidelines recommend that an IPF diagnosis requires the following:17
- Exclusion of other known causes of ILD (for example, domestic and occupational environmental exposures, connective tissue disease and drug toxicity) and either 2 or 3 below:
- The presence of a UIP pattern on high-resolution computed-tomography (HRCT)
- Specific combinations of HRCT and histopathological (surgical lung biopsy) pattern in patients subjected to surgical lung biopsy
Table 2. HRCT scanning patterns.17
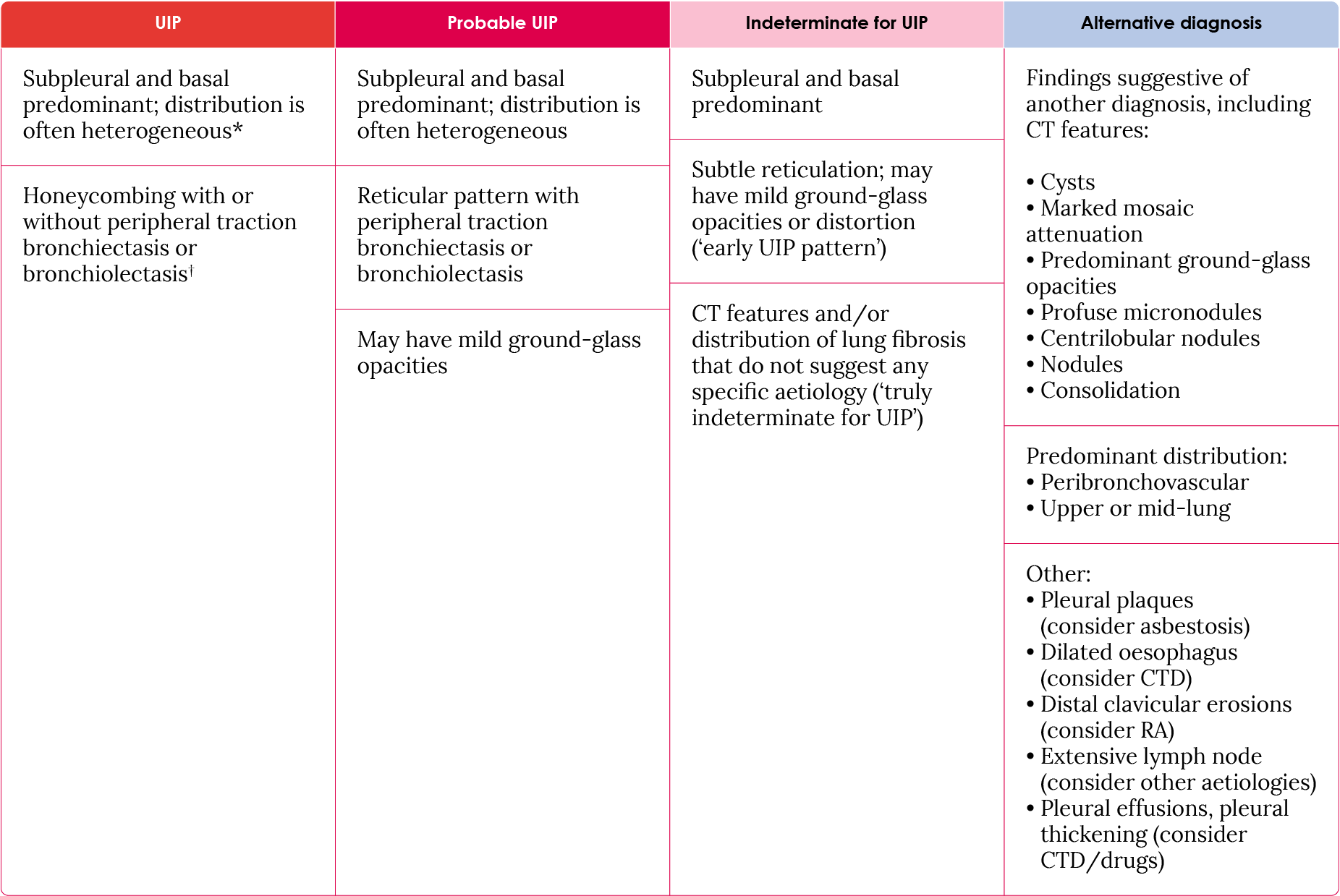
†Superimposed CT features: mild ground-glass opacities, reticular pattern, pulmonary ossification. *Variants of distribution: occasionally diffuse, may be asymmetrical.
CT, computed tomography; CTD, connective tissue disorders; HRCT, high-resolution computed-tomography; UIP, usual interstitial pneumonia; RA, rheumatoid arthritis.
Table 3. Histopathological UIP patterns and features17

*Granulomas, hyaline membranes (other than when associated with acute exacerbation of IPF, which may be the presenting manifestation in some patients), prominent airway-centred changes, areas of interstitial inflammation lacking associated fibrosis, marked chronic fibrous pleuritis, organizing pneumonia. Such features may not be overt or easily seen to the untrained eye and often need to be specifically sought. †Features that should raise concerns about the likelihood of an alternative diagnosis include a cellular inflammatory infiltrate away from areas of honeycombing, prominent lymphoid hyperplasia including secondary germinal centres, and a distinctly bronchiolocentric distribution that could include extensive peribronchiolar metaplasia.
IPF, idiopathic pulmonary fibrosis; UIP, usual interstitial pneumonia.
Table 4. Combinations of HRCT and histopathological (surgical lung biopsy) results required for an IPF diagnosis.17
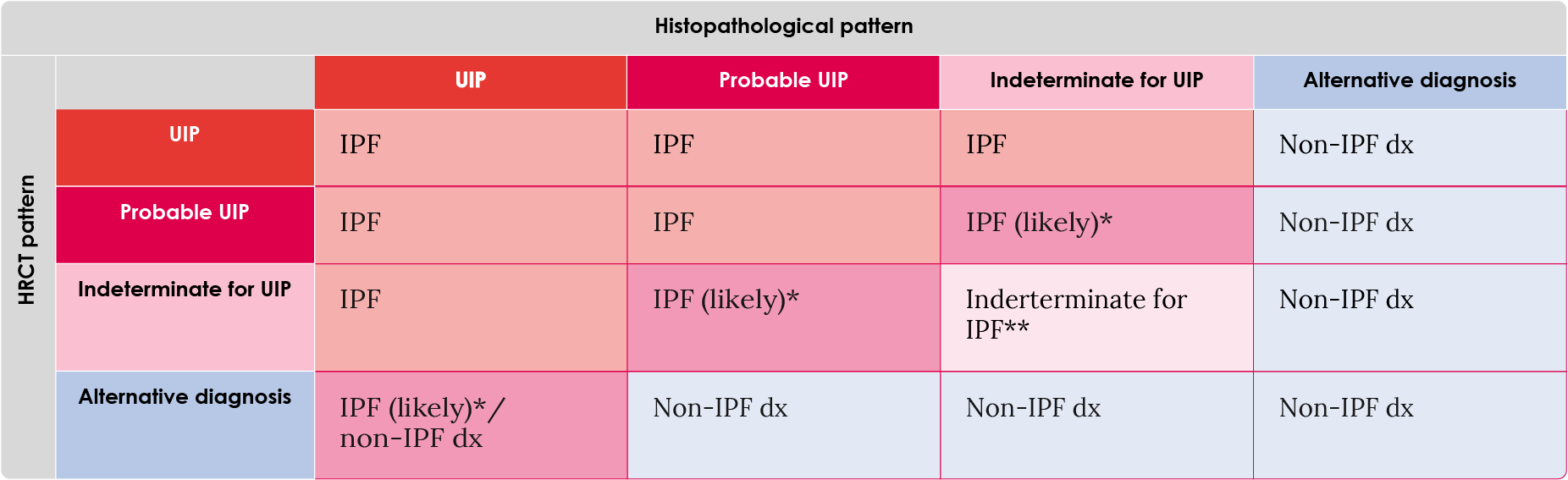
*IPF is the likely diagnosis when any of the following features are present:
- Moderate-to-severe traction bronchiectasis/bronchiolectasis (defined as mild traction bronchiectasis/bronchiolectasis in four or more lobes including the lingual as a lobe, or moderate to severe traction bronchiectasis in two or more lobes) in a man over age 50 years or in a woman over age 60 years
- Extensive (>30%) reticulation on HRCT and an age >70 years
- Increased neutrophils and/or absence of lymphocytosis in bronchoalveolar lavage fluid
- Multidisciplinary discussion reaches a confident diagnosis of IPF
** Indeterminate for IPF:
- Without an adequate biopsy is unlikely to be IPF
- With an adequate biopsy may be reclassified to a more specific diagnosis after multidisciplinary discussion and/or additional consultation
Dx, diagnosis; IPF, idiopathic pulmonary fibrosis; UIP, usual interstitial pneumonia.
Histopathology is also very useful for informing management of autoimmune ILD. Find out more in the video by Professor Kristin Highland: 'Do the results of histopathology alter management of autoimmune ILD?'
Diagnostic algorithm
The diagnosis of idiopathic pulmonary fibrosis (IPF) requires the exclusion of all known causes of fibrotic interstitial pneumonia and the presence of an idiopathic UIP pattern (Figure 5).17
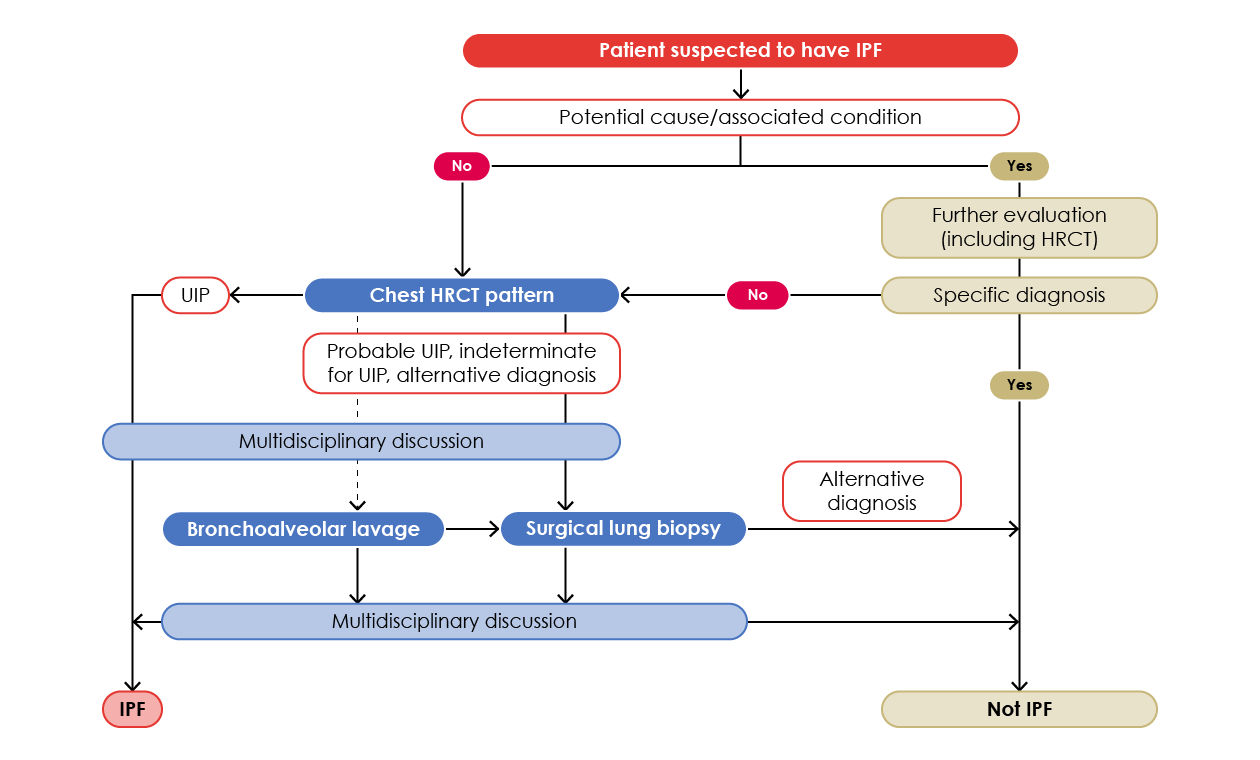
Figure 5. Diagnostic algorithm for IPF.17
We asked Professor Elisabeth Bendstrup whether the identification of genetic polymorphisms or other biomarkers would help improve timely diagnosis of IPF. Find out how she replied in the video 'Do you feel that the identification of genetic polymorphisms and other biomarkers will help improve the diagnosis of IPF? If so, how?'.
Prognosis
The natural history of idiopathic pulmonary fibrosis (IPF) is highly variable making the disease course in an individual patient difficult to predict (Figure 6).3
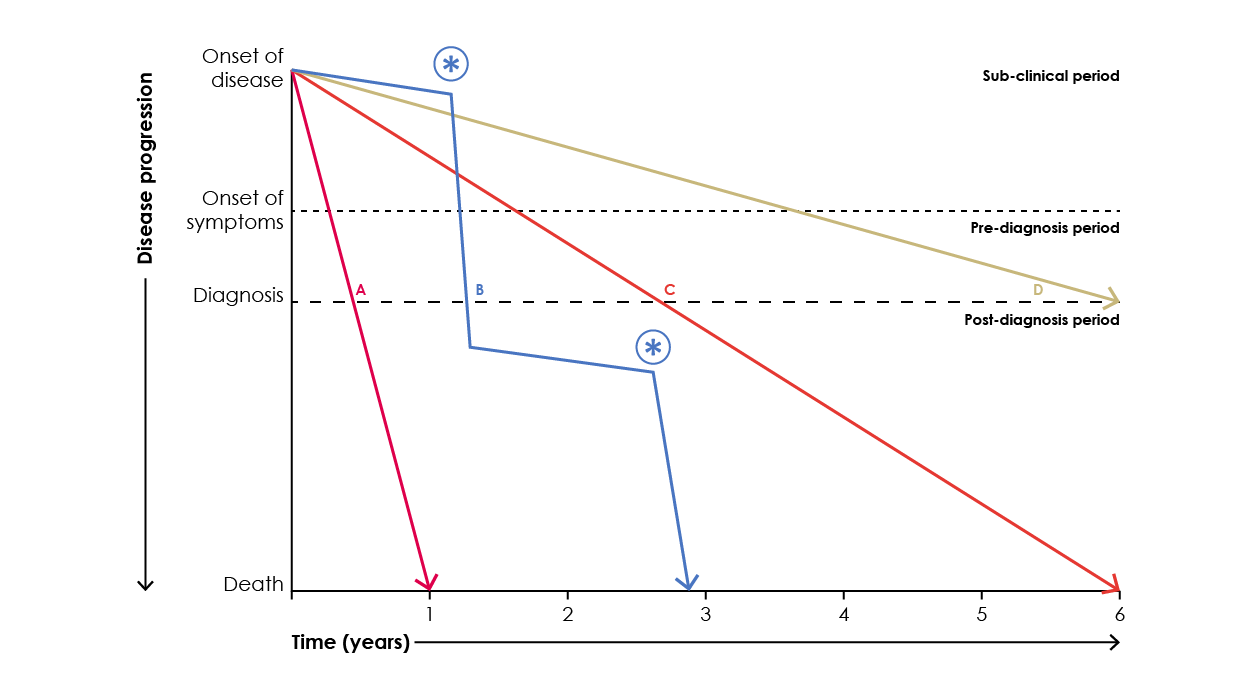
Figure 6. Possible clinical courses for patients with IPF.19
The rate of decline and progression to death may be rapid (line A), slow (lines C and D) or mixed (curve B), with periods of relative stability interrupted with periods of acute worsening (star).
Ultimately, IPF is an aggressive, fatal disease (median time for survival after diagnosis is two to three years), with most patients’ lung function gradually worsening over a period of years and a minority remaining stable or declining rapidly.2,18 Additionally, some patients may experience episodes of acute respiratory worsening despite previous stability.2
Evidence suggests that progressive fibrosis leads to gradual decline in pulmonary function: the placebo arms of several large, randomised controlled treatment trials in IPF demonstrate an average annual decline in forced vital capacity of approximately 0.2 litres in patients with mild-to-moderate pulmonary function abnormalities at the time of enrolment.2
Acute exacerbations
Between 5–10% of patients experience an acute exacerbation each year, which have a mortality rate of about 50% and are the main cause of hospitalisation and death in people with IPF.14
The intrinsic lung dysfunction underlying IPF may make patients more susceptible to external insults than healthy people. Indeed, as the figure below shows, many factors can trigger or increase the risk of an acute exacerbation:14,20,21
- Low or worsening FVC, low diffusing capacity for carbon monoxide
- Reduced six-minute walk distance, increased dyspnoea and poor oxygenation
- Comorbidities
- New ‘ground-glass’ opacities on computed tomography
- Respiratory viral infection, smoking (possibly)
- Thoracic surgery, lung biopsy and bonchoscopy
- Demographics (younger age, higher body mass index)
Comorbidities
Effective management of comorbidities improves the course of IPF and may increase survival.20 In a study of 272 patients (mean age 68.5 years; 76.5% male) a mean of 2.68 comorbidities was reported and the number of comorbidities was associated with reduced survival (Figure 7).22
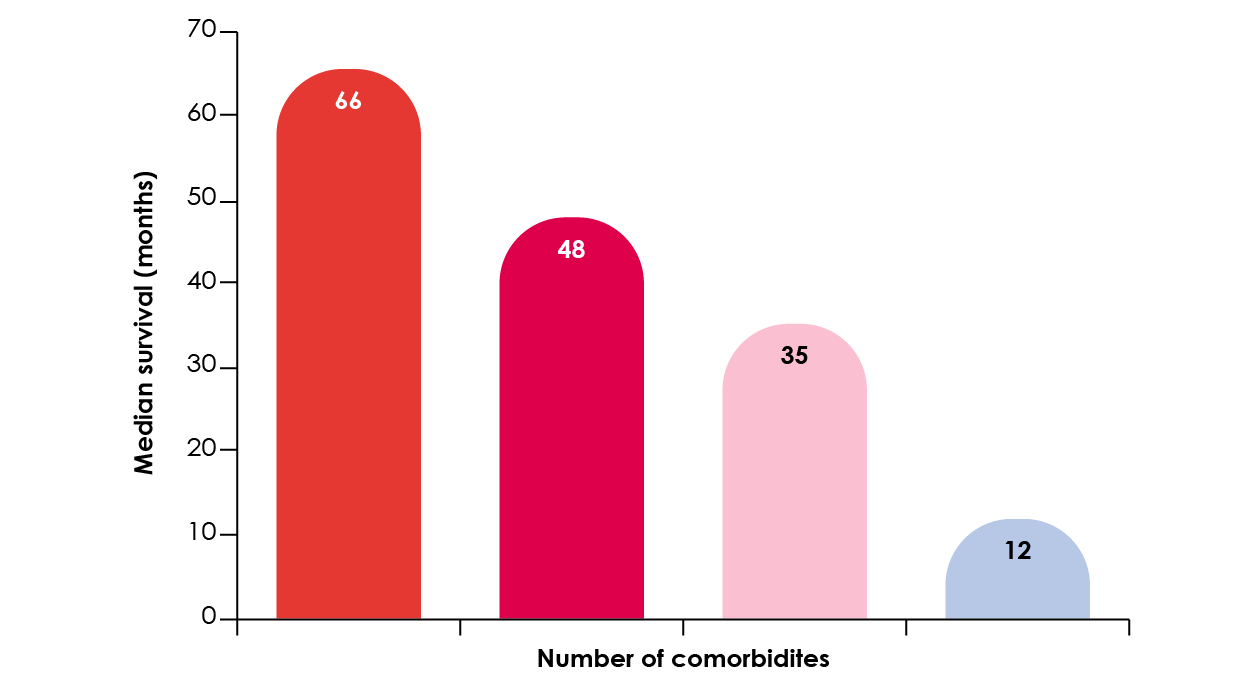
Figure 7. The relationship between number of comorbidities and survival.22
of interest
are looking at
saved
next event
of interest
are looking at
saved
next event
IPF References
1. King J, Costabel U, Cordier JF, DoPico GA, DuBois RM, Lynch D, et al. Idiopathic pulmonary fibrosis: diagnosis and treatment: International Consensus Statement. American Journal of Respiratory and Critical Care Medicine. 2000;161(2 I):646–664.
2. Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. An Official ATS/ERS/JRS/ALAT Statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824.
3. Sauleda J, Núñez B, Sala E, Soriano JB. Idiopathic Pulmonary Fibrosis: Epidemiology, natural history, phenotypes. Med Sci (Basel, Switzerland). 2018;6(4).
4. Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J. 2015;46(3):795–806.
5. Ley B, Collard HR. Epidemiology of idiopathic pulmonary fibrosis. Clinical Epidemiology. 2013. doi:10.2147/CLEP.S54815.
6. Collard HR, Chen SY, Yeh WS, Li Q, Lee YC, Wang A, et al. Health care utilization and costs of idiopathic pulmonary fibrosis in U.S. medicare beneficiaries aged 65 years and older. Ann Am Thorac Soc. 2015;12(7):981–987.
7. Van Manen MJG, Geelhoed JJM, Tak NC, Wijsenbeek MS. Optimizing quality of life in patients with idiopathic pulmonary fibrosis. Therapeutic advances in respiratory disease. 2017;11(3):157–169.
8. Glaspole IN, Chapman SA, Cooper WA, Ellis SJ, Goh NS, Hopkins PM, et al. Health-related quality of life in idiopathic pulmonary fibrosis: Data from the Australian IPF Registry. Respirology. 2017;22(5):950–956.
9. Swigris JJ, Kuschner WG, Jacobs SS, Wilson SR, Gould MK. Health-related quality of life in patients with idiopathic pulmonary fibrosis: a systematic review. Thorax. 2005;60:588–594.
10. Kreuter M, Bonella F, Wijsenbeek M, Maher TM, Spagnolo P. Pharmacological treatment of idiopathic pulmonary fibrosis: Current approaches, unsolved issues, and future perspectives. 2015. doi:10.1155/2015/329481.
11. Sgalla G, Iovene B, Calvello M, Ori M, Varone F, Richeldi L. Idiopathic pulmonary fibrosis: Pathogenesis and management. Respiratory Research. 2018;19(1). doi:10.1186/s12931-018-0730-2.
12. Wolters PJ, Collard HR, Jones KD. Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol. 2014;9:157–79.
13. Raghu G, Rochwerg B, Zhang Y, Garcia CAC, Azuma A, Behr J, et al. AMERICAN THORACIC SOCIETY An official ATS / ERS / JRS / ALAT clinical practice guideline: Treatment of idiopathic pulmonary fibrosis an update of the 2011 clinical practice guideline. 2015;192:3–19.
14. Kim HJ, Perlman D, Tomic R. Natural history of idiopathic pulmonary fibrosis. Respir Med. 2015;109(6):661–670.
15. de Boer K, Lee JS. Under-recognised co-morbidities in idiopathic pulmonary fibrosis: A review. Respirology. 2016;21(6):995–1004.
16. Molina-Molina M, Aburto M, Acosta O, Ancochea J, Rodríguez-Portal JA, Sauleda J, et al. Importance of early diagnosis and treatment in idiopathic pulmonary fibrosis. Expert Review of Respiratory Medicine. 2018;12(7):537–539.
17. Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. Diagnosis of idiopathic pulmonary fibrosis An Official ATS/ERS/JRS/ALAT Clinical practice guideline. Am J Respir Crit Care Med. 2018;198(5):e44–e68.
18. Cottin V, Cordier JF. Velcro crackles: The key for early diagnosis of idiopathic pulmonary fibrosis? European Respiratory Journal. 2012. doi:10.1183/09031936.00001612.
19. Ley B, Collard HR, King TE. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;183(4):431–440.
20. Glassberg MK. Overview of idiopathic pulmonary fibrosis, evidence-based guidelines, and recent developments in the treatment landscape. Am J Manag Care. 2019;25(11 Suppl):S195–S203.
21. Collard HR, Ryerson CJ, Corte TJ, Jenkins G, Kondoh Y, Lederer DJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An international working group report. Am J Respir Crit Care Med. 2016;194(3):265–275.
22. Kreuter M, Ehlers-Tenenbaum S, Palmowski K, Bruhwyler J, Oltmanns U, Muley T, et al. Impact of comorbidities on mortality in patients with idiopathic pulmonary fibrosis. PLoS One. 2016;11(3):e0151425.
23. Raghu G, Richeldi L. Current approaches to the management of idiopathic pulmonary fibrosis. Respir Med. 2017;129. doi:10.1016/j.rmed.2017.05.017.
24. King, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. A phase 3 trial of pirfenidone in patients with idiopathic Pulmonary Fibrosis. N Engl J Med. 2014;22:2083–92.
25. Richeldi L, Du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;22:2071–82.
26. Crestani B, Huggins JT, Kaye M, Costabel U, Glaspole I, Ogura T, et al. Long-term safety and tolerability of nintedanib in patients with idiopathic pulmonary fibrosis: results from the open-label extension study, INPULSIS-ON. Lancet Respir Med. 2019;7(1):60–68.
27. Nathan SD, Albera C, Bradford WZ, Costabel U, Glaspole I, Glassberg MK, et al. Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir Med. 2017;5(1):33–41.
28. Richeldi L, Cottin V, Du Bois RM, Es Selman M, Kimura T, Bailes Z, et al. Nintedanib in patients with idiopathic pulmonary fibrosis: Combined evidence from the TOMORROW and INPULSIS trials. Respir Med. 2016;113:74–79.
29. Wollin L, Wex E, Pautsch A, Schnapp G, Hostettler KE, Stowasser S, et al. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. European Respiratory Journal. 2015;45(5):1434–1445.
30. Lopez-de la Mora DA, Sanchez-Roque C, Montoya-Buelna M, Sanchez-Enriquez S, Lucano-Landeros S, Macias-Barragan J, et al. Role and new insights of pirfenidone in fibrotic diseases. International journal of medical sciences. 2015;12(11):840–847.
31. Takeda Y, Tsujino K, Kijima T, Kumanogoh A. Efficacy and safety of pirfenidone for idiopathic pulmonary fibrosis. Patient Preference and Adherence. 2014;8:361–370.
32. Ofev Prescribing Information. 2014. Available at https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/205832s000lbl.pdf (accessed September 2019).
33. Corte T, Bonella F, Crestani B, Demedts MG, Richeldi L, Coeck C, et al. Safety, tolerability and appropriate use of nintedanib in idiopathic pulmonary fibrosis. 2012. doi:10.1186/s12931-015-0276-5.
34. Kato M, Sasaki S, Nakamura T, Kurokawa K, Yamada T, Ochi Y, et al. Gastrointestinal adverse effects of nintedanib and the associated risk factors in patients with idiopathic pulmonary fibrosis. Sci Rep. 2019;9(1):12062.
35. Esbriet PI. Esbriet Prescribing Information. 2014.
36. Kolb M, Richeldi L, Behr J, Maher TM, Tang W, Stowasser S, et al. Nintedanib in patients with idiopathic pulmonary fibrosis and preserved lung volume. Thorax. 2017;72:340–346.
37. Vainshelboim B, Oliveira J, Yehoshua L, Weiss I, Fox BD, Fruchter O, et al. Exercise training-based pulmonary rehabilitation program is clinically beneficial for idiopathic pulmonary fibrosis. Respiration. 2014;88(5):378–388.
38. Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet. 2017;389(10082):1941–1952.
39. Flaherty KR, Fell CD, Huggins JT, Nunes H, Sussman R, Valenzuela C, et al. Safety of nintedanib added to pirfenidone treatment for idiopathic pulmonary fibrosis. Eur Respir J. 2018;52(2). doi:10.1183/13993003.00230-2018.
40. Vancheri C, Kreuter M, Richeldi L, Ryerson CJ, Valeyre D, Grutters JC, et al. Nintedanib with add-on pirfenidone in idiopathic pulmonary fibrosis: Results of the INJOURNEY trial. Am J Respir Crit Care Med. 2018;197(3):356–363.
41. Maher TM, van der Aar EM, Van de Steen O, Allamassey L, Desrivot J, Dupont S, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of GLPG1690, a novel autotaxin inhibitor, to treat idiopathic pulmonary fibrosis (FLORA): a phase 2a randomised placebo-controlled trial. Lancet Respir Med. 2018;6(8):627–635.
42. Maher TM, Kreuter M, Lederer DJ, Brown KK, Wuyts W, Verbruggen N, et al. Rationale, design and objectives of two phase III, randomised, placebo-controlled studies of GLPG1690, a novel autotaxin inhibitor, in idiopathic pulmonary fibrosis (ISABELA 1 and 2). BMJ Open Respir Res. 2019;6(1). doi:10.1136/bmjresp-2019-000422.
43. Raghu G, Scholand MB, De Andrade J, Lancaster L, Mageto Y, Goldin J, et al. FG-3019 anti-connective tissue growth factor monoclonal antibody: Results of an open-label clinical trial in idiopathic pulmonary fibrosis. Eur Respir J. 2016;47(5):1481–1491.
44. NCT03955146. Evaluation of Efficacy and Safety of Pamrevlumab in Patients With Idiopathic Pulmonary Fibrosis. 2019. Available at https://clinicaltrials.gov/ct2/show/NCT03955146?term=FG-3019&cond=Pulmonary+Fibrosis&rank=4 (accessed Septemer 2019).
45. Khalil N, Manganas H, Ryerson CJ, Shapera S, Cantin AM, Hernandez P, et al. Phase 2 clinical trial of PBI-4050 in patients with idiopathic pulmonary fibrosis. Eur Respir J. 2019;53(3).
46. Raghu G, Van Den Blink B, Hamblin MJ, Whitney Brown A, Golden JA, Ho LA, et al. Effect of recombinant human pentraxin 2 vs placebo on change in forced vital capacity in patients with idiopathic pulmonary fibrosis a randomized clinical trial. JAMA - J Am Med Assoc. 2018;319(22):2299–2307.
47. Raghu G, van den Blink B, Hamblin MJ, Brown AW, Golden JA, Ho LA, et al. Long-term treatment with recombinant human pentraxin 2 protein in patients with idiopathic pulmonary fibrosis: an open-label extension study. Lancet Respir Med. 2019;7(8):657–664.
This content has been developed independently by Medthority who previously received educational funding in order to help provide its healthcare professional members with access to the highest quality medical and scientific information, education and associated relevant content.