
Atopic Dermatitis overview
Are you aware of the recent updates on atopic dermatitis? Read below to:
- Engage with epidemiological images for atopic dermatitis across the globe
- Learn more about treatment options and the evidence behind them
- Find key detail relating to the presentation of atopic dermatitis and burden of the disease
Atopic dermatitis epidemiology
Prevalence and incidence of atopic dermatitis
Atopic dermatitis, or atopic eczema, is the most common chronic skin condition and is characterised by acute episodes of eczematous, pruritic lesions over dry skin. It typically starts in early childhood and is widely reported to affect 15–20% of children and 1–3% of adults1.
Since the 1970s, the incidence of atopic dermatitis has increased 2- to 3-fold in industrialised nations2.
While atopic dermatitis affects a substantial number of infants and adults, the severity can differ significantly between patients. Studies have shown reasonable heterogeneity in the relative levels of severity observed in patients. Analysis of the 2007–2008 National Survey of Children’s Health found that 67% of US children reported mild disease, 26% reported moderate disease and 7% reported severe disease3,4. However, a smaller study in the UK where disease severity was assessed by a dermatologist rather than patient self-report found that 84% of children had mild disease, 14% had moderate disease and just 2% had severe disease5.
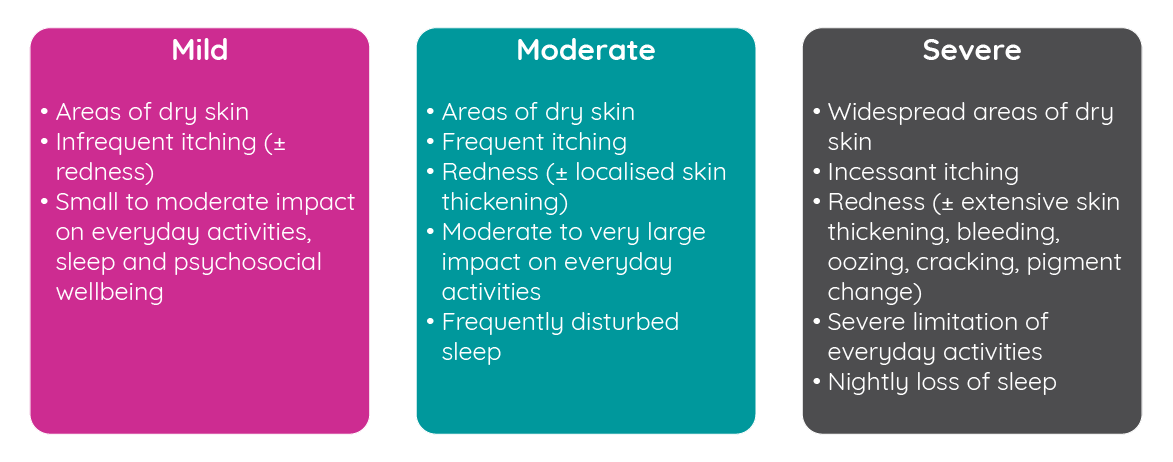
Figure 1: Atopic dermatitis disease severity classification (adapted from the National Institute for Health and Care Excellence6).
In children, 60% develop the disease in the first year of life and 90% within the first 5 years of life7. It is also believed to represent the first step of the ‘atopic march’ towards other allergic conditions later in life such as asthma or allergic rhinitis1. In a 3-year, double-blind study of 1,091 infants (3–18 months of age) with recent onset atopic dermatitis (≤3 months), development of one or more atopic comorbidities was observed in 37.0% of infants at the end of observation (mean 2.8 years)8.
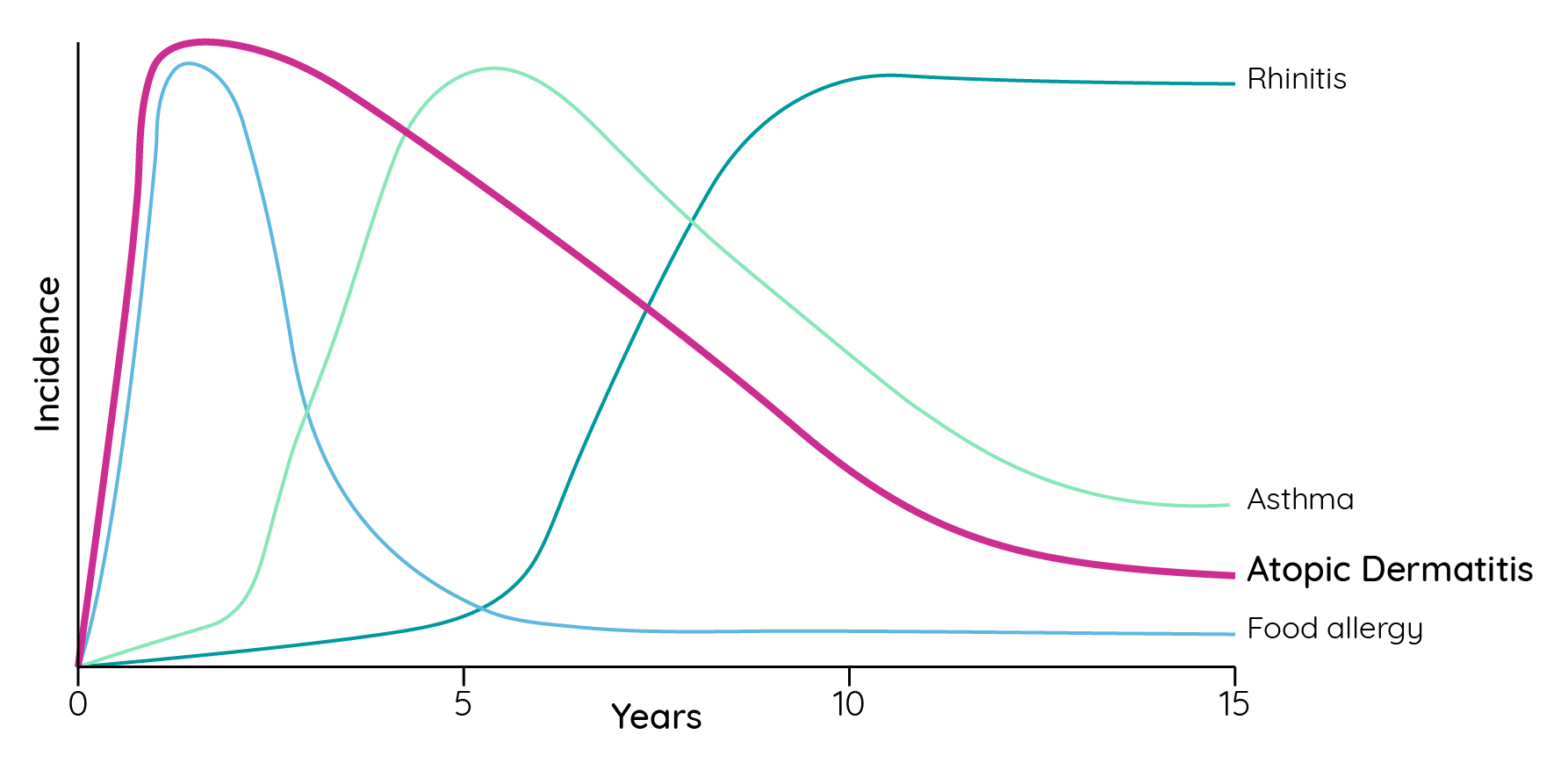
Figure 2: Incidence of different atopic diseases in childhood and adolescence (adapted from Barnetson & Rogers9).
In 70% of patients with childhood atopic dermatitis a spontaneous remission before adolescence is achieved1. Interestingly, however, a prospective cohort study of children with mild-to-moderate atopic dermatitis reported that 80% of patients with >5 years of follow-up continued to have symptoms or use medication. Furthermore, as patients aged, they sought healthcare support less frequently potentially influencing the common perception of the frequency with which atopic dermatitis resolves over time10. However, conflicting data and ongoing debate continues on this topic indicating that more research is needed to fully understand the long-term impact of atopic dermatitis11,12,13.
Although prevalence figures of 15–20% in children and 1–3% in adults are commonly quoted1, the American Academy of Dermatology guidelines suggest a childhood prevalence of up to 25%7.
Irrespective of the given prevalence, all these figures hide significant geographic and population variations. In the International Study of Asthma and Allergies in Childhood (ISAAC), a global study of 2 million children in 100 countries, prevalence in children aged 6–7 years was shown to be as low as 0.9% in parts of India and as high as 22.5% in Ecuador14.
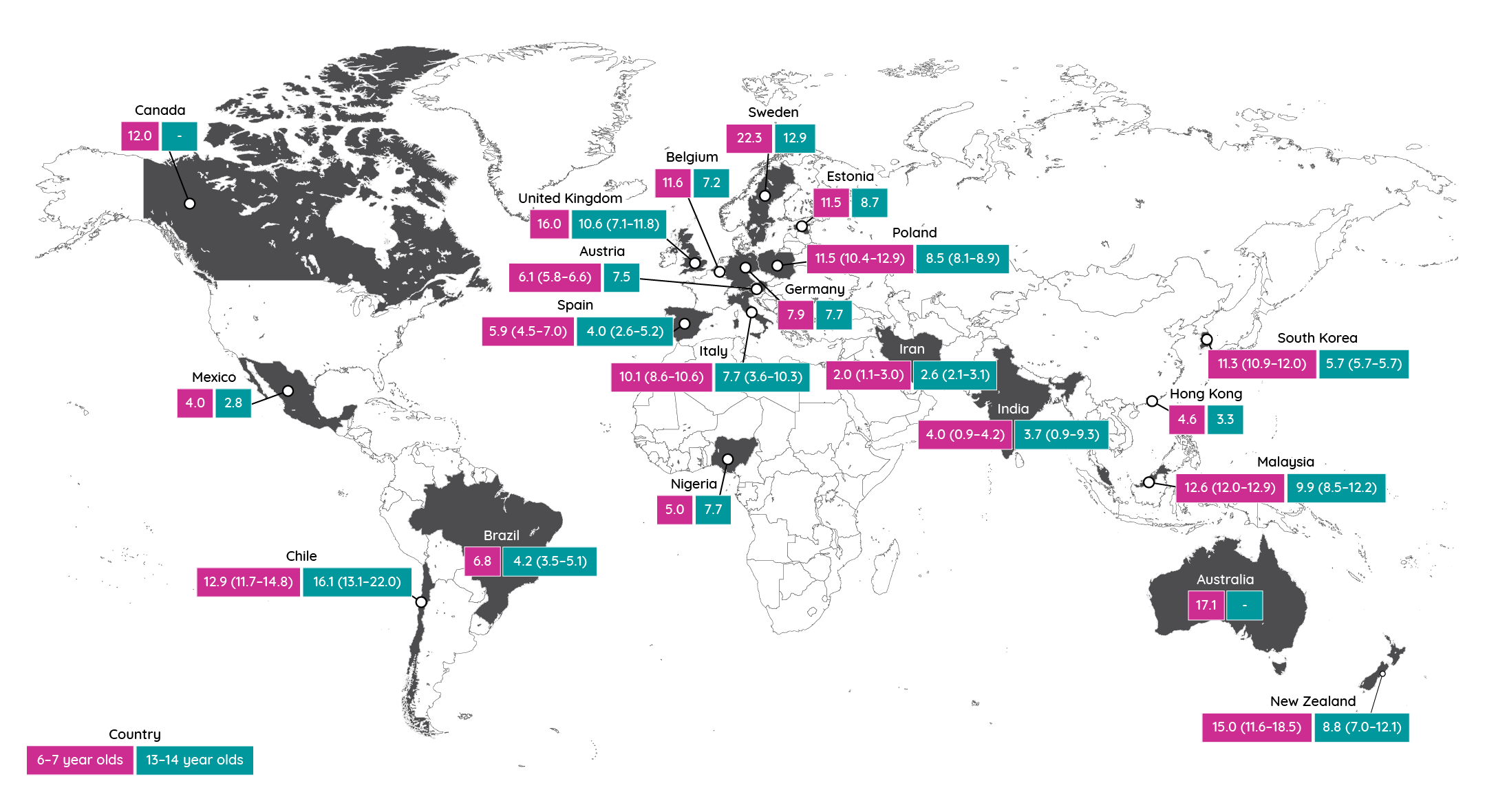
Figure 3: Prevalence of atopic dermatitis in the last 12 months in phase 3 of the ISAAC study (adapted from Williams et al.15). Figures in parentheses represent the range of prevalence observed in different regions of the country where available.
The different phases of the ISAAC study (phase 1 in early- to mid-1990s; phase 3 in early 2000s) have also enabled them to establish how the prevalence of atopic dermatitis is changing over time15.
In children aged 13 to 14 years old, the prevalence of atopic dermatitis in countries with very high levels has plateaued or decreased although the burden continued to rise in developing country settings. However, in children aged 6 to 7 years old, most countries showed an increase of two standard errors or more in atopic dermatitis over a 5- to 10-year period15. Meanwhile, in the USA, the National Health Interview Survey revealed that the prevalence of atopic dermatitis among children increased from 8% in 1997 to 12% in 2010–2011. However, it appears to have subsequently plateaued in 2012–201316.
While the ISAAC study provides valuable insight into the prevalence of childhood atopic dermatitis, are similar geographic disparities observed in the prevalence of adult disease? The European Community Respiratory Health Survey reported point prevalence rates that ranged from 0.3% (Switzerland) to 6.2% (Estonia)17. Furthermore, a series of US studies have identified point prevalence rates ranging from 3.2% to 10.7% depending on the population assessed and the definitions used18,19,20. A recent international, cross-sectional, web-based survey sought to address these gaps in understanding and received over 90,000 responses from people in the USA, Canada, France, Germany, Italy, Spain, the UK and Japan. Among these respondents, the overall 12-month prevalence of adult atopic dermatitis was 4.9% (95% CI 4.6%, 5.2%). Regional variability was seen in many countries, but a higher point prevalence was observed in Southern European countries than elsewhere (<0.05)21.
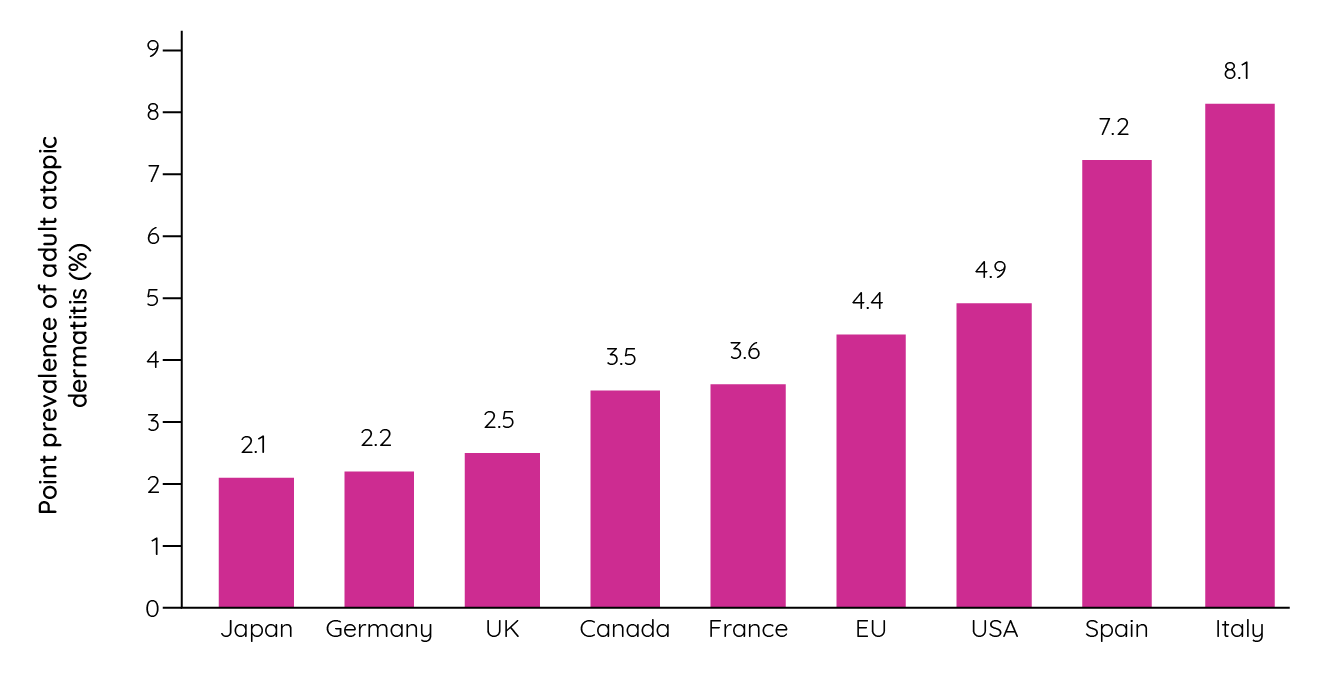
Figure 4: Point prevalence of adult atopic dermatitis by country (adapted from Barbarot et al.21).
Ethnicity and atopic dermatitis
While gender does not appear to affect the risk of developing atopic dermatitis, race and ethnicity have been implicated7,22. Studies have suggested that atopic dermatitis is more commonly observed in Asian and black individuals than white. The ISAAC study suggested that people in Africa and Oceania have a higher rate of atopic dermatitis than people from the Indian subcontinent and Northern/Eastern Europe14. Population studies in the USA support these insights with African American children having a higher prevalence of atopic dermatitis than European American children. In fact, one study suggested that after adjusting for a number of environmental and societal variables, African American children are 1.7 times more likely to develop atopic dermatitis than European American children23. Similar results were seen in a UK-based study where 16.3% of London-born black Caribbean children were diagnosed with atopic dermatitis versus 8.7% of white children24.
Aetiology of atopic dermatitis
Atopic dermatitis is a complex, multifactorial disease with a host of genetic and environmental factors contributing to its development.
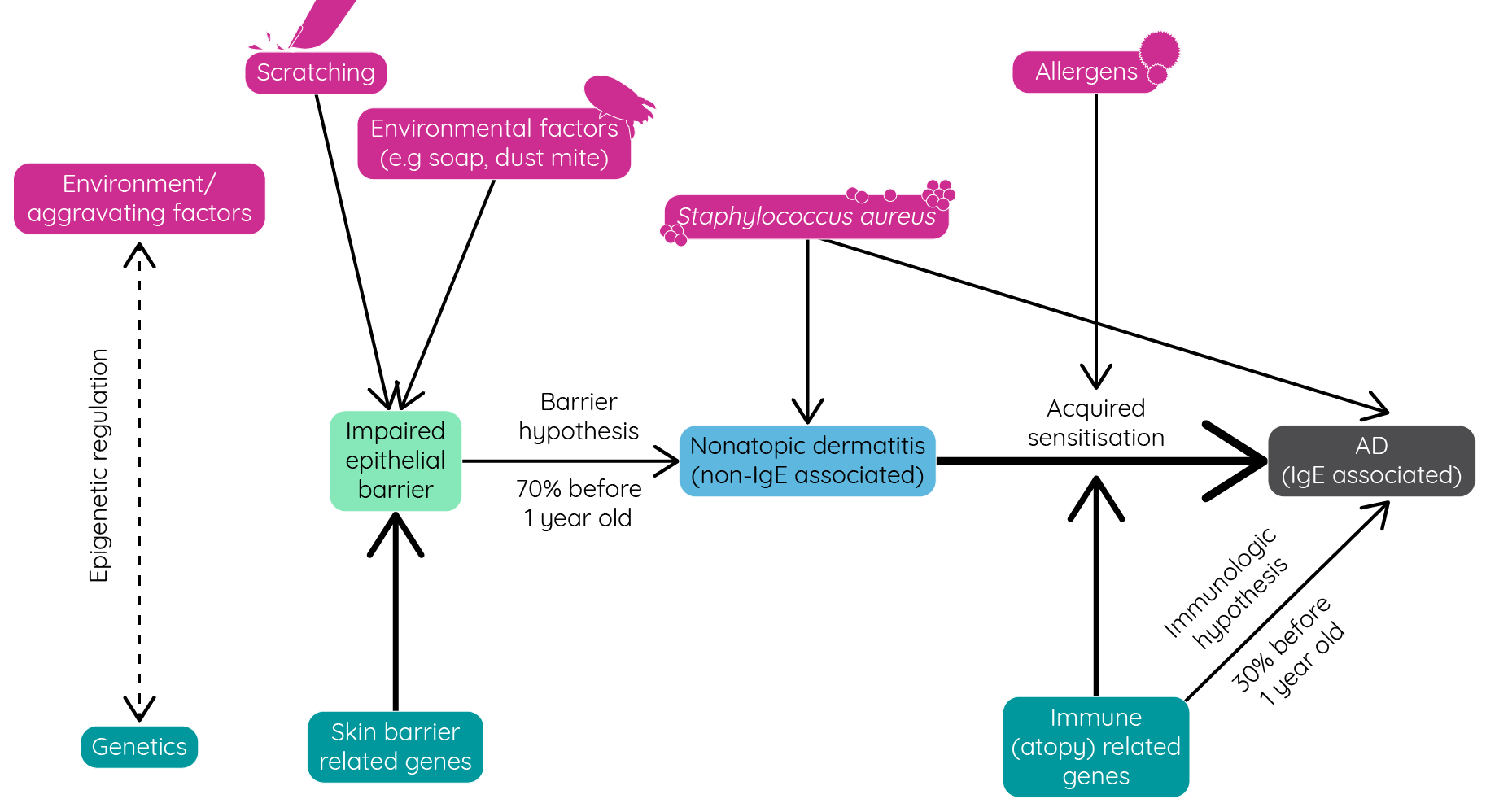
Figure 5: Genetic and environmental factors affecting the development of atopic dermatitis (adapted from Nutten et al.1).
Genetic susceptibility
A family history of atopic disease strongly correlates with atopic dermatitis with around 70% of patients with atopic dermatitis having family members with atopic disease. In fact, the risk of atopic dermatitis increases by 2- to 3-fold when one parent has a history of atopic disease and by 3- to 5-fold when it is both parents2.
Subsequent genetic analyses have identified several chromosomal regions linked to the development of atopic dermatitis. These include genes on chromosome regions 1q21 and 5q31 to 5q332. In addition, more than 100 published articles have identified over 80 genes with a possible association with atopic dermatitis25.
Filaggrin
Filaggrin is a major structural protein of the stratum corneum and plays a crucial role in skin barrier integrity22. The FLG gene, which encodes the filaggrin protein, is located on chromosome 1q21.3 and several loss-of-function mutations have been identified1. Patients with atopic dermatitis carrying a FLG loss-of-function mutation exhibit an earlier onset of disease, more severe disease, a higher rate of eczema herpeticum, and a greater incidence of other atopic diseases versus those with wildtype FLG22.
Approximately 10% of white Europeans are heterozygous carriers of a FLG loss-of-function mutation, however, this increases to about 50% of European atopic dermatitis patients1,2. However, this falls to 27% of Asian patients and is rare in African patients with no association at all observed in an Ethiopian study22. These studies suggest that while FLG mutations are associated with the development of atopic dermatitis, the fact that they are absent in many patients and that immune-mediated skin inflammation is similar between patients with and without FLG mutations, other genes are also likely to play a role22.
Interleukins
Several interleukins have been identified as potentially playing a role in the pathophysiology of atopic dermatitis. The chromosome region 5q31 to 5q33 has been linked to atopic dermatitis development and within this region are the genes for IL-4, IL-5, IL-12, IL-13 and granulocyte-macrophage colony stimulating factor (GM-CSF). These cytokines can influence atopic dermatitis by upregulating IgE production and reducing the expression of proteins involved in skin barrier function, including filaggrin2,26.
Skin flora
The skin of patients with atopic dermatitis is frequently colonised by Staphylococcus aureus, which can exacerbate or aggravate skin lesions27. The cause of this may be reduced production of antimicrobial peptides that form part of the innate immune system and are typically found on the skin. These have been seen to be reduced in patients with atopic dermatitis and may be the reason that they are more susceptible to infection28. Interestingly, a recent study showed that the skin of African American patients with atopic dermatitis was typically colonised by different strains of S. aureus than European American or Mexican American patients (who were shown to have similar strains on their skin). This may go on to influence the presentation and infection types seen in different patient populations29.
Environmental factors
A range of environmental factors have been identified that affect the risk of developing atopic dermatitis. Some of these, such as climate variations, can be seen through the differing prevalence rates reported between countries and geographic regions.
Substantial variations in prevalence, nationally and internationally, suggest that environmental and genetic factors are the main drivers of change in the disease burden1.
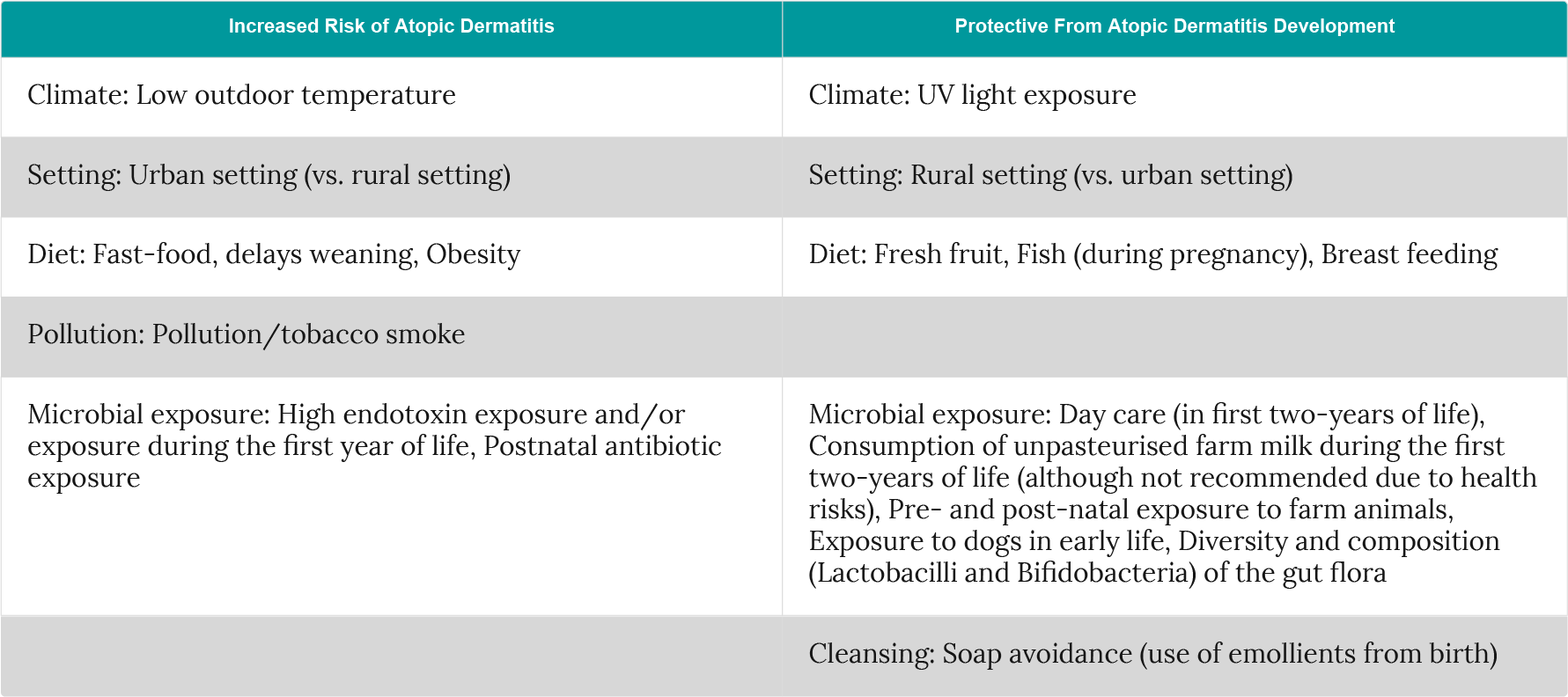
These environmental factors can influence skin barrier function while microbial exposure may influence immune sensitisation1. The below table outlines several environmental risk factors that potentially influence atopic dermatitis development although some of the data remains inconclusive (Table 1).
Table 1: Selected environmental risk- and protective-factors for atopic dermatitis (adapted from Nutten et al.1; Eichenfield et al.7; Simpson et al.30; Glatz et al.31).
Triggers of atopic dermatitis
Atopic dermatitis can be triggered by a range of mechanisms, and trigger avoidance should be encouraged to help control disease activity. Triggers for atopic dermatitis include:
- Viral infections
- Food allergens
- Cosmetics
- Fragrances
- Extremes of hot and cold weather
- Wool
- Environmental allergens (e.g., dust mites, pollens, moulds, tobacco smoke, animal dander).
Some controversy persists with the role of food allergens in the pathogenesis of atopic dermatitis. Around 50–70% of children with early-onset atopic dermatitis are sensitive to one or more allergens, with these typically being a food allergen2,1. In infancy, food allergens contribute to approximately 40% of atopic dermatitis cases, but do not cause it2. Instead, food allergies are co-associated with, or an exacerbating factor, for atopic dermatitis1.
Atopic dermatitis pathophysiology
A complex range of factors influence the development and onset of atopic dermatitis, but what is the underlying mechanism that drives skin barrier dysfunction and inflammation? Discover how breakdown of the skin barrier can lead to allergen penetration and immune system activation.
Professor Seemal Desai describes current key hypotheses and research areas regarding the specific mechanisms involved in the pathophysiology of atopic dermatitis.
Pathogenesis
In the past, atopic dermatitis was thought to be primarily a consequence of immune dysfunction in susceptible children. However, recent advances point towards a complex pathophysiology with genetic, barrier function, immunity and environmental factors acting together and synergistically to drive barrier dysfunction, inflammation and disease progression25.
Two theories have been proposed for the pathophysiology of atopic dermatitis. The inside-out model proposes that an allergic response triggers inflammation that disrupts the skin barrier enabling further allergen and microbe exposure. The outside-in hypothesis, however, suggests that skin barrier dysfunction initiates the process of atopic dermatitis, with immune dysregulation occurring subsequently2.
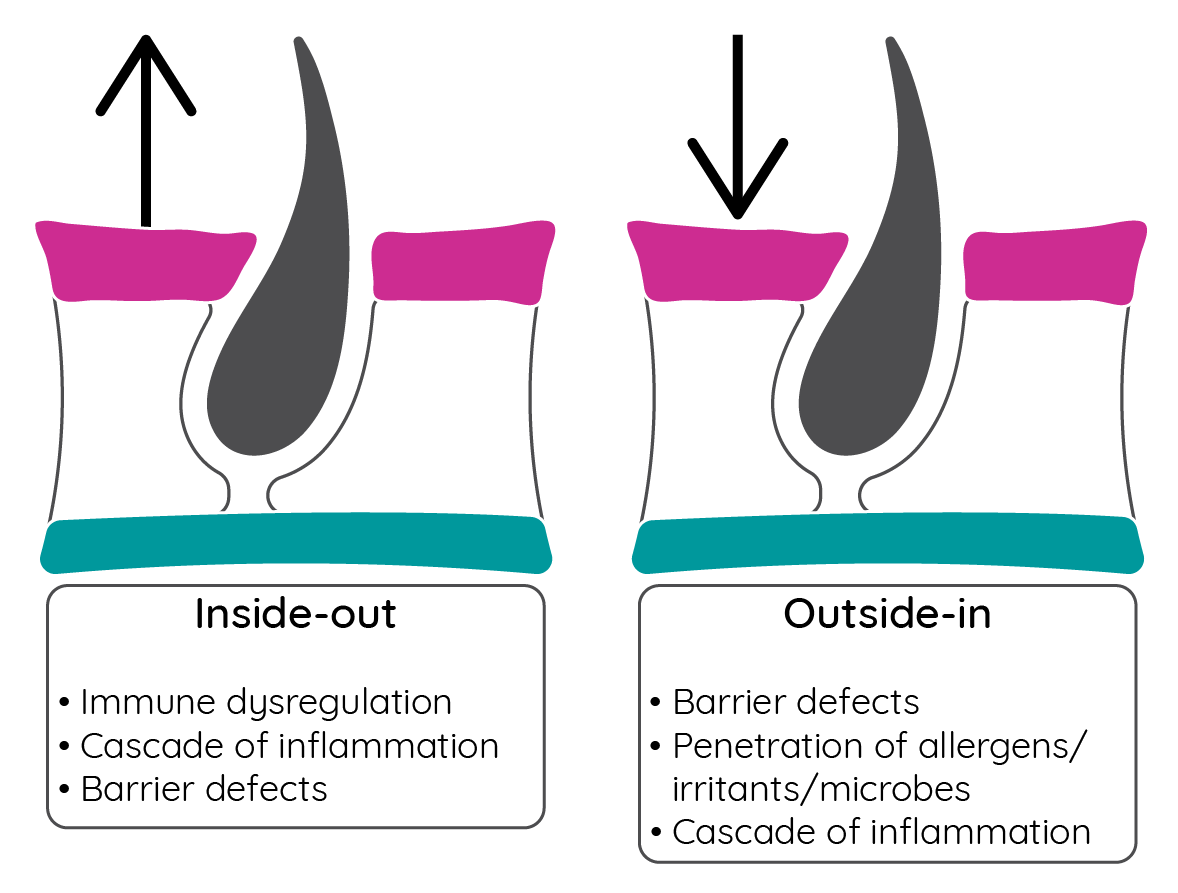
Figure 6: Theories of atopic dermatitis pathogenesis (adapted from Sidbury & Barton32).
While some suggest that these models are unlikely to be exclusive and probably both play a role2, others suggest that skin barrier dysfunction, or the outside-in model, is the initiating step26.
Skin barrier dysfunction and atopic dermatitis
Within the epidermis, barrier proteins including filaggrin, transglutaminases, keratins and loricrin, and intercellular proteins cross-link into clusters to form an impermeable barrier26. This protects us from exogenous stressors and allergens, but also helps maintain internal fluid and electrolyte homeostasis. Disruption of the skin barrier enables the penetration of allergens that can trigger an immune response and may subsequently lead to the development of IgE-mediated allergies25. While skin barrier dysfunction is critical to the onset of atopic dermatitis, no individual cause has been identified. Instead, several factors contribute to the loss of skin barrier function.
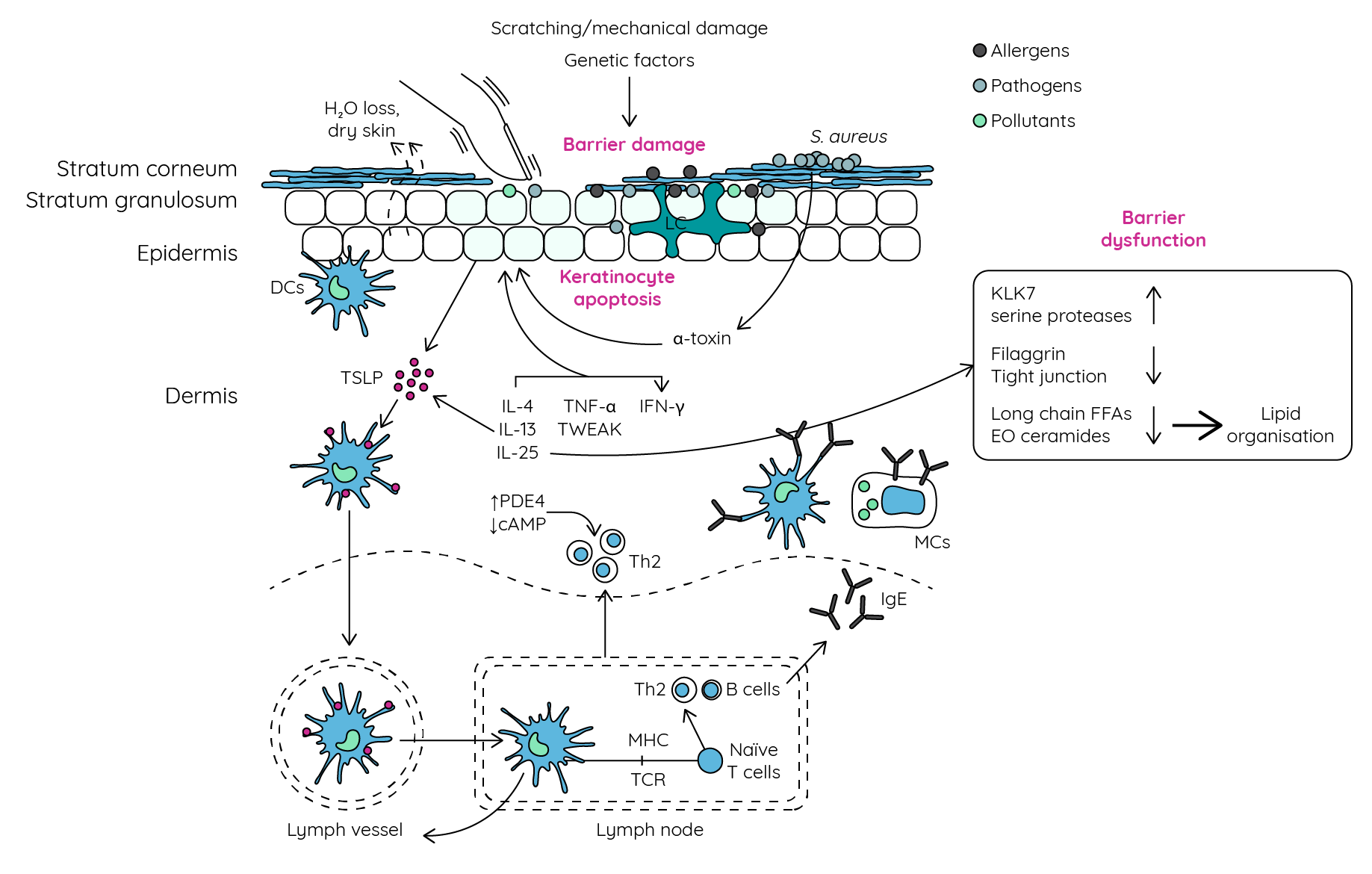
Figure 7: Skin barrier dysfunction and atopic dermatitis (adapted from Peng & Novak33; Guttman-Yassky et al.34). Genetic, immunological, and mechanical factors lead to skin barrier dysfunction allowing activated antigen-presenting cells to encounter allergens, pathogens, and environmental factors. This results in promotion of a Th2-mediated immune response further damaging the skin barrier and inducing keratinocyte apoptosis. Increases in PDE4 also increase Th2 cytokine expression. Virulence factors released by colonising pathogens can also promote keratinocyte cell death and Th2-type inflammation further driving barrier dysfunction. DC, Dendritic cell; EO ceramides, ester-linked ω-hydroxy ceramides; FFAs, free fatty acids; IFN-γ, interferon-γ; IgE, immunoglobulin E; IL, interleukin; KLK7, kallikrein 7; LC, Langerhans cell; MCs, mast cells; MHC, major histocompatibility complex; PDE4, phosphodiesterase-4; TCR, T cell receptor; Th2, T helper 2 cells, TNFα, tumour necrosis factor α; TSLP, thymic stromal lymphopoietin; TWEAK, TNF-like weak inducer of apoptosis.
Epidermal barrier protein loss
While filaggrin plays an important role in the development of barrier protein clusters, it is also degraded upon exposure to low humidity into free amino acids, which are essential for maintaining skin pH and retaining water in the cornified layer35,36,37,38. Mutation of the filaggrin gene, FLG, is a well-known predisposing factor for atopic dermatitis development.
Filaggrin deficiency has been shown to cause paracellular skin barrier abnormalities that reduce inflammatory thresholds to irritants and haptens26. However, a significant number of patients with atopic dermatitis do not have a FLG mutation and approximately 40% of individuals with FLG-null alleles do not go on to develop atopic dermatitis39,26.
While FLG mutations clearly contribute to the development of atopic dermatitis in many people, it is not sufficient alone to generate the condition. Interestingly, FLG mutation is not the only means by which filaggrin levels are reduced.
A Th2-type inflammatory response is typically observed in acute atopic dermatitis and the Th2 cytokines IL-17, IL-22, IL-25 and IL-31 are capable of downregulating filaggrin expression. Indeed, while filaggrin mutations are observed in about one-third of Caucasian patients with atopic dermatitis, skin barrier impairment and reduced filaggrin expression are seen in most patients33.
Tight junctions, desmosomes and adherens junctions form adhesions between the cells of the epidermis to help create a physical, permeability barrier. The Th2 cytokines that downregulate filaggrin expression have also been shown to downregulate the proteins involved in tight junction formation resulting in increased transepidermal water loss, greater allergen and microbial penetration of the skin and reduced skin barrier cohesion40,41,26.
Antimicrobial peptides (AMPs) and dysbiosis
During infection, inflammation and wounding, keratinocytes express a range of AMPs that form an innate chemical barrier to microbes. However, the presence of Th2 cytokines in atopic dermatitis downregulates their expression and has been associated with an increased risk of S. aureus infection, which can further exacerbate atopic dermatitis26. Cutaneous dysbiosis can cause skin inflammation and barrier dysfunction and result in recurrent infection and increased skin pH42,43,44.
Lipid matrix
The presence of S. aureus as well as Th2 cytokines has also been shown to modify the expression of enzymes involved in the biosynthesis of long-chain free fatty acids and ceramides within the skin further impacting skin barrier function45,46,47,48.
Keratinocyte cell death and damage
With skin barrier function reduced, the subsequent immune response can exacerbate dysfunction and promote physical damage. Increased expression of TNFα and TNF-like weak inducer of apoptosis (TWEAK) promotes keratinocyte apoptosis and development of skin lesions49. In addition, the keratinocytes of patients with atopic dermatitis have been shown to be more susceptible to IFN-γ-induced apoptosis compared to healthy controls50. Further damage to the skin barrier can be caused by scratching. Pruritus is a hallmark feature of atopic dermatitis, however, scratching further irritates the skin exacerbating the itching7.
The immune system in atopic dermatitis
As skin barrier function decreases, allergen stimulation is believed to result in the release of thymic stromal lymphopoietin (TSLP), which stimulates dendritic cells to promote a Th2-driven immune response by promoting differentiation of naïve T cells into Th2 cells51,52,53. As the inflammatory pathway progresses, an increased infiltration of T cells, dendritic cell subtypes, macrophages, mast cells and eosinophils within the skin is observed54,53. Patients with atopic dermatitis have a genetically determined dominance of Th2 lymphocytes55 and this translates into increased levels of the Th2 cytokines IL-4, IL-13, IL-25 and IL-3333.
The key cytokines within a Th2-driven immune response are IL-4 and IL-13. Both cytokines signal via the IL-4 receptor α (IL-4Rα) to promote IgE production by B cells, dendritic cell differentiation, activation of T cells and recruitment of eosinophils33. Interestingly, the immune cells that infiltrate the skin of atopic dermatitis patients might behave differently to those seen in individuals without the condition. Activation of Toll-like receptor 2 (TLR2) on monocytes and macrophages from patients with atopic dermatitis resulted in a different cytokine and chemokine response compared to those from healthy controls56,57,58.
The key cytokines within a Th2-driven immune response are IL-4 and IL-13. Both cytokines signal via the IL-4 receptor α (IL-4Rα) to promote IgE production by B cells, dendritic cell differentiation, activation of T cells and recruitment of eosinophils33.
Mast cells also play an important role in the inflammatory response as well as being key mediators of type 1 hypersensitivity. The number of mast cells is elevated in the skin lesions of patients with atopic dermatitis and it has been suggested that they promote skin inflammation in this setting through the release of IL-3159. Furthermore, they have been shown to degranulate and release proinflammatory mediators following stimulation by δ toxin released by S. aureus60.
As atopic dermatitis progresses towards a chronic state, the underlying inflammatory response changes and shifts from predominantly Th2-driven inflammation to mixed Th2/Th1/Th17/Th22 response. The consequences of this change are greater keratinocyte apoptosis due to Th1-mediated release of IFN-γ and tissue remodelling, increased skin thickness and lichenification as a result of Th22 cell activation33.
The role of phosphodiesterase-4 in atopic dermatitis
Many phosphodiesterase (PDE) enzymes are known to be involved in atopic and inflammatory processes61. Notably, PDE4 plays a role in regulating the inflammatory cascade associated with atopic dermatitis.
PDE4 inhibition, resulting in increases in cyclic AMP (cAMP) levels, is linked to the suppression of proinflammatory molecules such as IL-4 and prostaglandin E234.
Typically, cAMP is present at high levels within a variety of cells with PDE4 regulating its breakdown34. However, it has been shown that patients with atopic dermatitis display elevated levels of PDE4 in circulating inflammatory cells, resulting in a reduction in cAMP and an increase in proinflammatory cytokine production62. PDE4 levels in dermal fibroblasts have also been observed to be increased in patients with atopic dermatitis63.
High levels of PDE4 activity can lead to increased prostaglandin E2 production, resulting in increased levels of IL-1064. IL-10 is normally considered to be an inhibitor of inflammation by reducing cytokine production65. However, in atopic dermatitis monocytes, IL-10 causes a decrease in the production of IFNγ by Th1 cells leading to a change in the Th1/Th2 ratio towards a Th2 response64. This regulation of Th1 and Th2 functional responses can account for various characteristics of atopic dermatitis such as: increases in the production of IL-4, IL-5, and IL-6 by T cells, increased IgE synthesis, decreased interferon-γ production and impaired cell-mediated immune response64. As a consequence, inhibiting the PDE4 enzyme has become an important therapeutic target for reversing inflammatory abnormalities in atopic dermatitis66.
To learn more about this, visit our treatment section focused on the role of PDE4 inhibitors and their mechanism of action.
Role of the JAK-STAT signalling pathway in atopic dermatitis
The JAK STAT pathway is activated by the key inflammatory mediator IL-4. The role of JAK-STAT, which includes members of both the JAK and STAT family, is important in atopic dermatitis due to the effect that it has on the regulation of Th2 differentiation. Complex pathways mediate this effect. Upregulation of the TH2 immune response by the JAK-STAT pathway results in excessive release of many different cytokines, angiogenic factors and chemokines, and the rise of IgE. This is able to bind to mast cells increasing the process of inflammation in atopic dermatitis. The JAK-STAT pathway can also cause changes in the barrier function of the skin. As some JAK inhibitors (oral and topical) have demonstrated a positive effect on the severity of atopic dermatitis and symptoms experienced, they are currently undergoing further testing and exploration for safety and efficacy. One example is abrocitinib, an oral selective inhibitor of JAK-1, currently being investigated for its safety and efficacy in the treatment of moderate to severe atopic dermatitis. There remain questions as to whether selective JAK-1 versus non-selective JAK inhibitors may give the better safety profile67.
Pruritis in atopic dermatitis
Problematic pruritus in atopic dermatitis
Itch is the most important clinical symptom in atopic dermatitis and has been shown to have a profound impact on the quality of life of both child and adult patients68,69. Importantly, chronic pruritus affects nearly everyone with atopic dermatitis with a point prevalence of 87–100%70,71.
"Without adequate therapy that addresses both the pathophysiological and emotional component of atopic dermatitis-related itch, problems with disease management and adherence to therapy amass in children, putting these patients at increased risk for the long-term sequelae associated with paediatric atopic dermatitis"68.
What are the underlying mechanisms of pruritis in atopic dermatitis?
Watch our expert Professor Martin Steinhoff introduce the signals and mediators involved in pruritis in atopic dermatitis:
Histamine, involved primarily in acute itch, is not alone in mediating pruritis. Having said this, because histamine is a well-known factor causing itch with urticaria caused by the degranulation of mast cells, antihistamine have been commonly used as a treatment. However, it has been found that because many other mechanisms are involved in the pathophysiology of the condition, antihistamines are not fully effective as a management strategy72.
Understanding itch in atopic dermatitis
It is apparent that histaminergic (as seen in urticaria) and non-histaminergic (such as atopic dermatitis) mechanisms can drive pruritus. While closely related, these two mechanisms appear to be independent of one another. In the skin, both mechanisms of pruritus have their own receptors and cutaneous nerve fibres, which continues into the central nervous system (CNS) where each has its own specialised tracts and neural structures73. Chronic pruritus, as seen in atopic dermatitis, is induced by the nonhistaminergic pathway and is likely to be a consequence of multiple factors related to skin barrier dysfunction.
Transepidermal water loss (TEWL) increases following skin barrier dysfunction due to a reduction in keratinocyte retention of water and increased loss of water from the surrounding epidermal environment74. Increased TEWL has been associated with itch intensity and has also been shown to increase the pH of the skin resulting in activation of serine proteases that promote further pruritus75,76. In fact, in atopic dermatitis, the receptor for proteases is typically upregulated and sensitised73.
Skin barrier dysfunction is also likely to allow increased entry of irritants and pruritogens, which further enhance the itch process. Upon entry to the skin, these compounds are capable of stimulating one of myriad receptors on the keratinocyte cell surface channels (such as PAR2, TRPV ion channels, IL-31 receptor, opioid receptors), promoting the release of pruritogenic molecules (including opioids, proteases, substance P, nerve growth factor, neutrotrophin 4, endocannabinoids). In addition, keratinocytes can release acetylcholine directly stimulating sensory nerves as well as lowering their threshold for stimuli activation73. Keratinocytes, therefore, can be stimulated by, and secrete, pruritic factors as well as communicate the itch sensation downstream.
Managing itch is multifactorial with a need to address the itch itself, but also contributing factors such as dry skin, inflammation, and scratch lesions. Basic therapy should be undertaken to address skin barrier dysfunction and includes the use of emollients, bath oils and avoiding any clinically relevant allergens. Beyond basic therapy, a range of topical and systemic treatment options are often used to help manage the itch associated with atopic dermatitis69.
Cytokines in itch
Cytokines are critical components in the pathogenesis of atopic dermatitis with some also playing an important role in atopic itch.
IL-2
Intradermal injection of IL-2 into patients with atopic dermatitis or healthy controls induced itching and erythema for 48–72 hours77. This link between IL-2 and itch has also been observed in patients receiving intravenous IL-2 as a cancer therapy with severe pruritus a recognised side effect78,79,80. Calcineurin promotes IL-2 synthesis and secretion80 and calcineurin inhibition with ciclosporin A was shown to reduce pruritus in patients with atopic dermatitis81.
IL-4 and IL-13
IL-4 and IL-13 help promote the Th2-driven immune response observed in patients with atopic dermatitis. Levels of both cytokines are increased in lesional skin82 and IL-13 levels are elevated in the serum of patients with atopic dermatitis where they correlate with disease severity83. The role of IL-4 and IL-13 in pruritus has been confirmed using a monoclonal antibody that targets the action of both IL-4 and IL-13 (dupilumab), which reduced pruritus severity by over 50% during clinical trials84.
IL-5
The Th2 cytokine IL-5, acts within inflamed tissue to attract eosinophils and has been identified as playing a key role in eosinophilic asthma. Mepolizumab is an anti-IL-5 monoclonal antibody that has been approved for the treatment of severe eosinophilic asthma. While initial studies in patients with atopic dermatitis were underwhelming85, studies are ongoing looking at the benefits of anti-IL-5 therapy in patients with an increased eosinophil count86.
IL-31
IL-31 expression is elevated in several pruritic conditions including atopic dermatitis, prurigo nodularis and allergic contact dermatitis87,88. It signals by binding to its receptor complex, expressed on keratinocytes, epithelial cells and by dorsal root ganglion pruriceptors, promoting transcription via the JAK/STAT pathway87,89,80. Interestingly, IL-31 has been shown to promote a late onset itch (mean delay 143 minutes) suggesting that it acts via indirect mechanisms90.
IL-33
IL-33 is a promoter of Th2-mediated inflammation, a key driver of atopic dermatitis91,92,93. Elevated levels have been observed in the lesional skin and serum of patients with atopic dermatitis where serum levels correlated with disease severity94. Little is known about how IL-33 is involved in chronic pruritus, however, recent in vitro data indicates that it promotes Mast cell expression and secretion of IL-31. Interestingly, IL-33-mediated upregulation of IL-31 was enhanced by co-stimulation with IL-4, substance P and IgE73,95.
IL-33 has recently been indicated to have a key link with pruritis. Professor Seemal Desai discusses this further.
Thymic stromal lymphopoietin (TSLP)
TSLP is an important mediator of T cell maturation and activation and is believed to be a key driver of atopic dermatitis initiation and maintenance96,97,98. Its expression is upregulated in keratinocytes in atopic dermatitis99 helping to promote Th2-mediated inflammation. TSLP is also a pruritogenic molecule that can promote itch directly via cutaneous sensory neurons97.
The itch-scratch cycle
The pruritic sensation drives a desire to scratch, and this in itself can lead to skin damage and the impairment of its barrier function. This dysfunction can lead to production of many of the cytokines and other inflammatory mediators, further exacerbating the unwanted stimulation of nerve fibre endings. As this happens, the brain comes to receive additional triggers to make an individual want to itch, continuing the itch-scratch cycle72. Professor Steinhoff explores this in more detail.
There are a number of known causes of the impulse to itch. It has been suggested that the mechanisms which drive itch can be classified as pruritoceptive (also referred to as dermatological), neurogenic (also referred to as systemic), neuropathic, and psychogenic100.
The pruritoceptive aetiology of itch refers to cellular processes and inflammatory mediators and pruritogens released by and affecting the skin. Pruritogens implicated in the pathology of atopic dermatitis within the category of pruritoceptive itch include histamine, tryptase cytokines and substance P100.
Over 90% of afferent fibres transmitting impulses to the brain are polymodal and can respond to at least two stimuli types. It has also been demonstrated through the stimulation of the skin that distinct nerve fibres respond to the histamine-mediated and non-histamine mediated itch, indicating that there are separate peripheral pathways enabling the transmission of impulses for different pruritogens101,102. It has been suggested that the scratch response to itch, which evolutionally may have been or be beneficial through its use to remove harmful elements from the outer layer of the body, may cause localised pain therefore supressing the itch sensation, and/or induce the release of serotonin, resulting in the perceived sensation of pleasure103,104. Of course, repeated scratching is not beneficial, aggravates the sensation of pruritis, and can lead to the chronic manifestation of this condition.
Professor Steinhoff tells us more about the management of pruritis in relation to the complex mechanisms by which the sensation of itch arises.
Atopic dermatitis symptoms
Atopic dermatitis, also called atopic eczema or eczema, is an inflammatory, chronic or chronically relapsing skin disease with pruritus as the predominant dermatological symptom68,105,69.
The condition typically presents in infancy, with 60% of cases occurring within the first year, however, it can develop in older patients with approximately one third of adult cases developing in adulthood68,69. Over time, the clinical presentation of atopic dermatitis changes68.
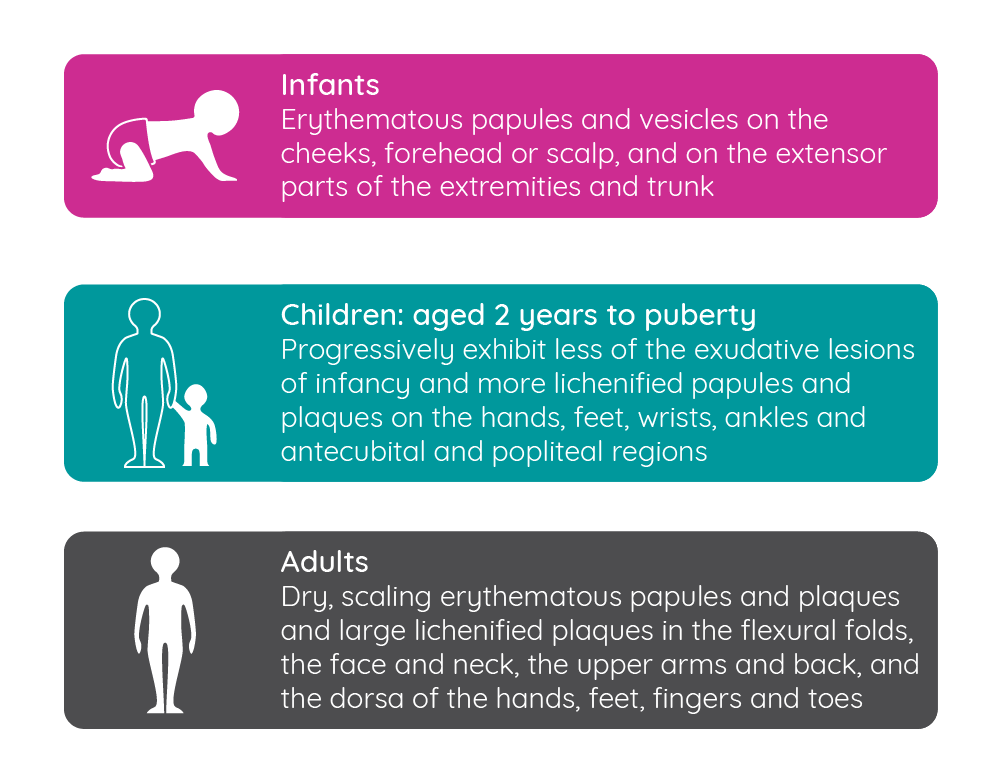
Figure 8: Changes in the clinical presentation of atopic dermatitis over time (adapted from Blume-Peytavi et al.68)
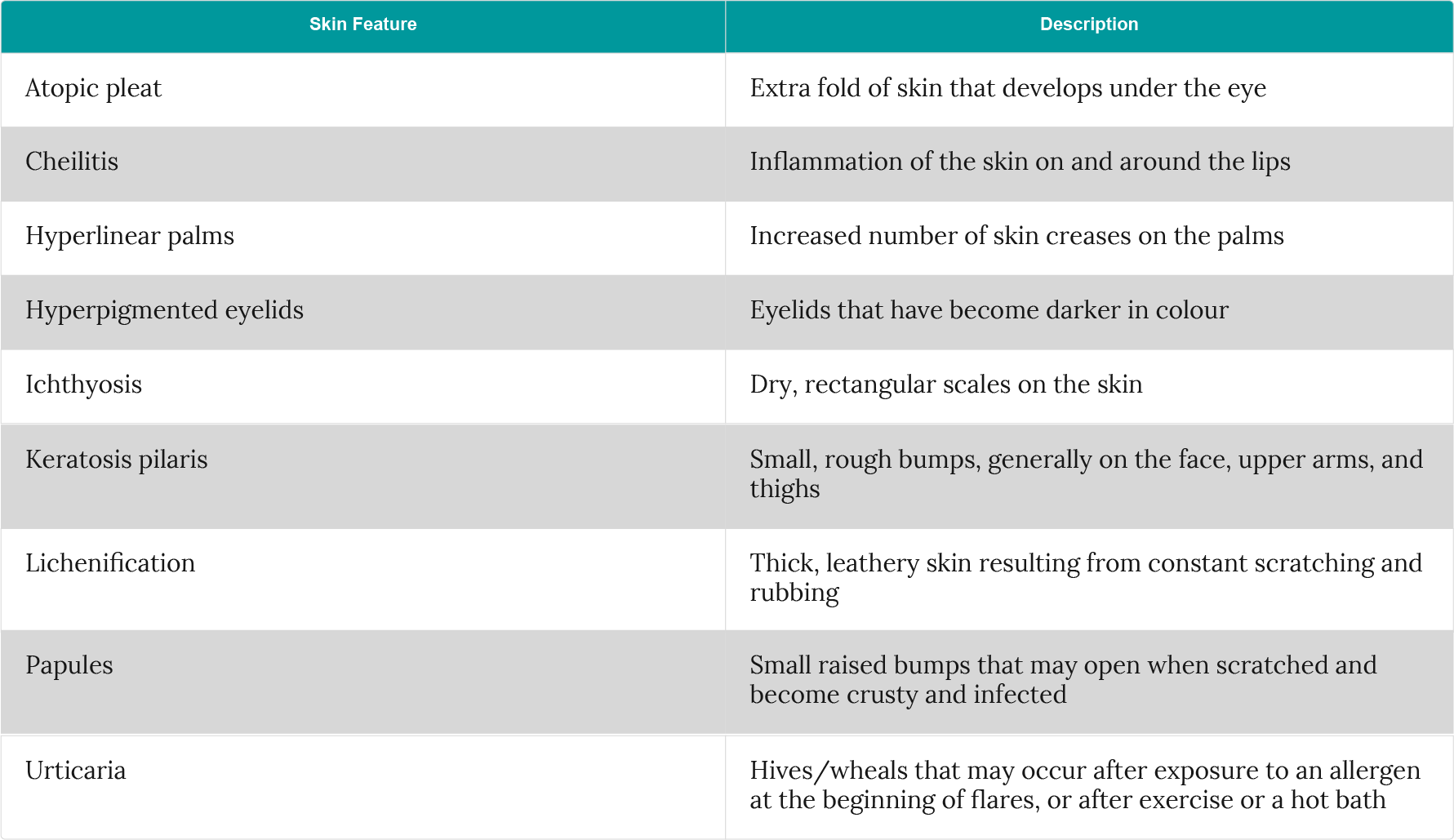
Skin features that are commonly associated with atopic dermatitis include:
Table 2: Common skin features associated with atopic dermatitis (adapted from Avena-Woods et al.2)
A recent prospective study of 356 adults with atopic dermatitis found that 41.9% of cases were adult-onset, with 24.4% developing after the age of 50 years. While many features of atopic dermatitis were similar between child and adult patients, such as body surface area affected and the Eczema Area and Severity Index, adult-onset atopic dermatitis was found to have distinct phenotypes with a lesional predilection for the hands and/or the head and neck106.
While the symptoms of atopic dermatitis may spontaneously resolve in patients, it is not curable. Visit our treatment goals section to find out more about the importance of effective treatment for atopic dermatitis.
Burden of disease for atopic dermatitis
As a chronic, relapsing skin condition, atopic dermatitis has a significant impact on patients’ quality of life. However, given the age of most patients, and the fact that it can persist into adulthood, atopic dermatitis also places a heavy burden on patients’ family as well as society2.
The misery of living with atopic dermatitis cannot be overstated for it may have a profoundly negative effect on the HRQoL of children and their family unit in many cases.
In fact, skin and subcutaneous diseases were the fourth leading cause of nonfatal burden in 2013 Global Burden of Disease Study, with dermatitis (atopic, seborrheic and contact) having the largest burden of that group107. Furthermore, a study using the Children’s Life Quality Index (CLQI) among children (aged 5–16 years) with various chronic diseases found generalised eczema to be second to only cerebral palsy in impact on quality of life108.
Dr Amy Paller discusses the urgent unmet needs that remain for patients with atopic dermatitis
Symptoms and comorbidities in atopic dermatitis
Pruritus, or itch, is the predominant symptom associated with atopic dermatitis. In a US study of 304 patients with atopic dermatitis, 91% experienced itch daily71. The impact of persistent itch should not be underestimated as it has been shown to affect both physical and emotional/psychological quality of life (QoL) domains. Indeed, reducing itch was identified as the most important treatment goal in 36% of patients with atopic dermatitis109. Furthermore, the physical damage caused by repeated scratching can exacerbate skin barrier breakdown and promote establishment of chronic disease68.
What is the burden of atopic dermatitis for patients?
Watch Professor Steinhoff discuss the considerable burden that pruritis can add for patients living with atopic dermatitis.
More recently, skin pain has also been identified as an important symptom associated with the burden of atopic dermatitis. A prospective, questionnaire-based study of patients with atopic dermatitis aged 13 years and older (N=305) established that 42.7% of patients had experienced skin pain in the past week, with 13.8% reporting it to be severe or very severe. Importantly, patients with severe itch and pain had significantly worse QoL scores than patients with only one or neither symptom110.
Asthma and allergic rhinitis
With atopic dermatitis frequently representing the first step in a series of allergy-related conditions (atopic march), it is well established that patients with atopic dermatitis have an increased risk of developing asthma and allergic rhinitis. In children with severe atopic dermatitis, as many as 50% went on to develop asthma and 75% developed allergic rhinitis111 from 14.2% to 52.7% with an odds ratio of developing asthma of 2.14 compared to children without atopic dermatitis. However, this may be higher than the risk for the general population given many of the included studies were performed in the hospital setting112. Interestingly, an Australian longitudinal study showed that childhood atopic dermatitis increased the risk of persistent allergic asthma in adulthood, but not non-allergic asthma offering potential therapeutic insights for these patients113.
Rheumatoid arthritis and inflammatory bowel disease
Gene mapping studies in atopic dermatitis have identified a number of loci involved in immune function that have also been implicated in other immune-mediated inflammatory diseases114. A large retrospective cohort study identified 49,847 individuals (N = 655,815; prevalence of 7.6%) under the age of 40 years who had prevalent atopic dermatitis between 2005 and 2006. After adjusting for age and sex, patients with atopic dermatitis, compared with those without atopic dermatitis, had significantly increased risks of Crohn’s disease, ulcerative colitis and rheumatoid arthritis115.
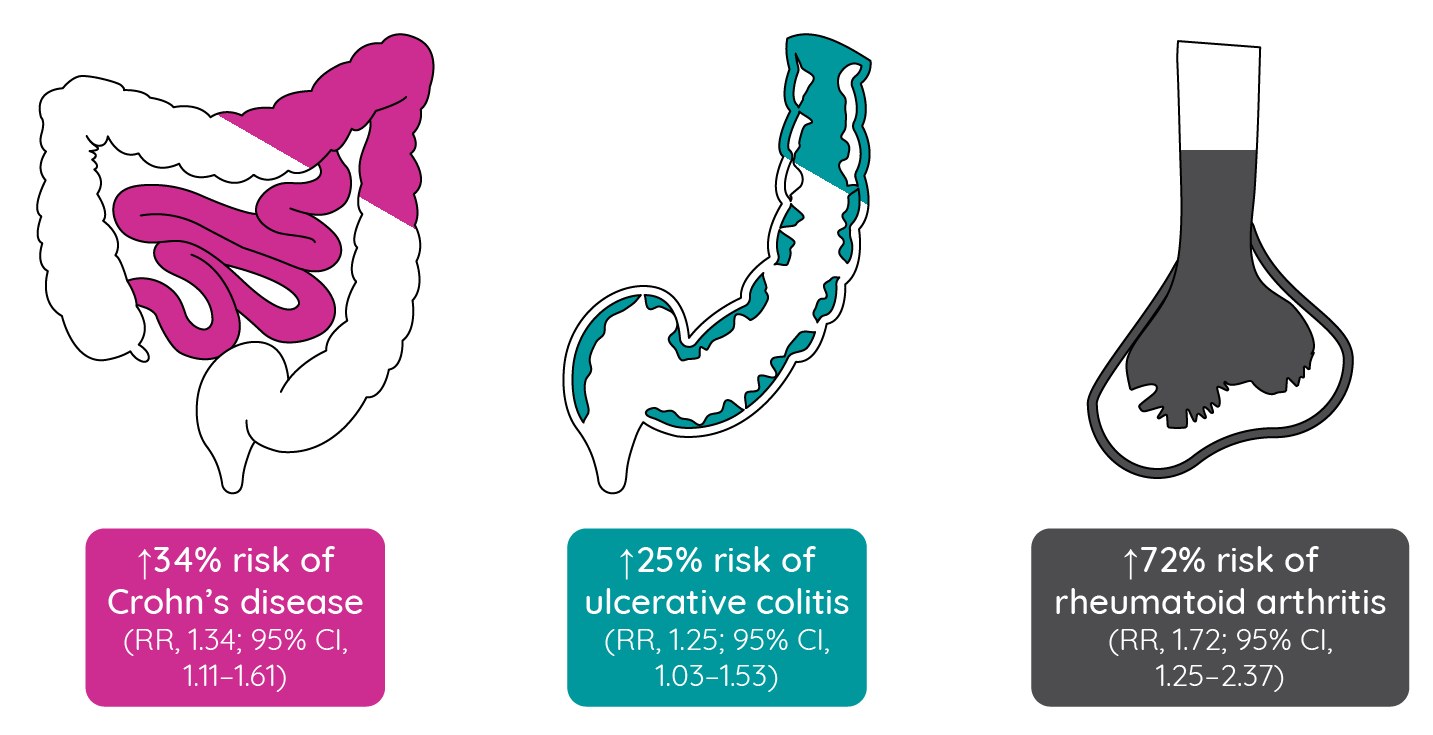
Figure 9: Risks of inflammatory bowel disease and rheumatoid arthritis comorbidities in patients with atopic dermatitis (adapted from Schmitt et al.115).
While the cause of these associations remains to be confirmed, it has been established that chronic atopic dermatitis, rheumatoid arthritis and inflammatory bowel disease all involve prominent Th1 and Th17 immune responses. It is tempting to speculate that sustained Th1/Th17 inflammation in patients with chronic atopic dermatitis leads to an increased risk of other chronic inflammatory conditions115.
Burden of childhood atopic dermatitis
Sleep disturbance is a significant problem for infants and has been associated with a range of daytime behavioural deficits116,117,118,119. Over 60% of children with atopic dermatitis experience disturbed sleep patterns with rates of up to 89% in children during exacerbations120,18,121. Furthermore, a U.S. population-based study found that children with atopic dermatitis had sleep disturbance at least 4 nights a week3.
Is the treatment goal different when comparing paediatric and adult patients?
Behavioural problems
Characterised by poor initiation, frequent awakenings and prolonged nocturnal wakefulness, disturbance to sleep in children with atopic dermatitis may result in more than the expected daytime fatigue122,123. Sleep disturbance has been linked to daytime behavioural problems including reduced child and family quality of life, increased discipline problems and attention deficit hyperactivity disorder (ADHD)122.
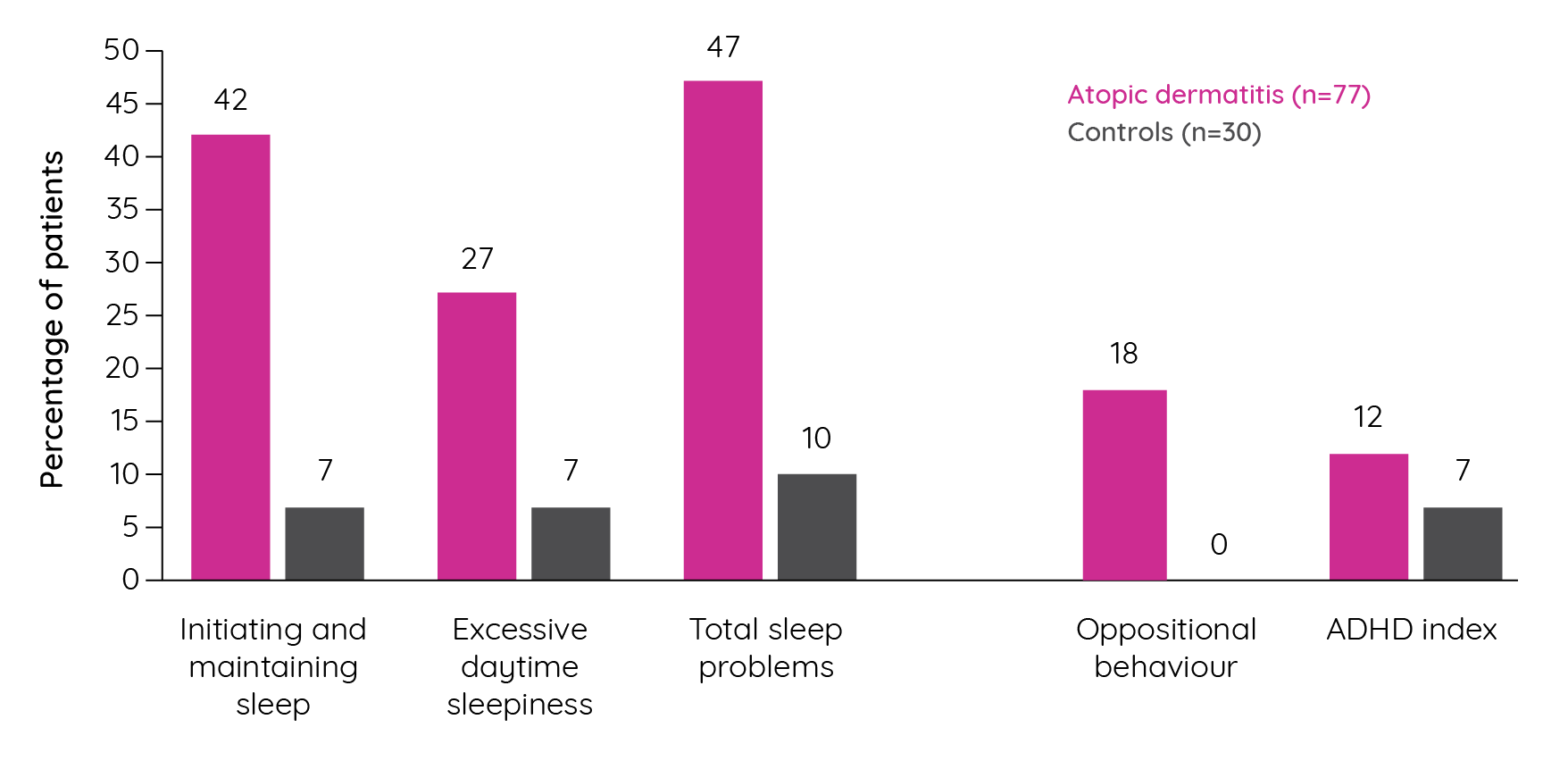
Figure 10. Percentage of children above the clinical cut-off for selected sleep and behavioural issues (adapted from Camfferman et al.122).
Subsequent confirmatory factor analyses indicated that asthma, atopic dermatitis and allergic rhinitis have a significant effect on childhood behaviour with it being substantially, but not completely, mediated by their impact on sleep122. Population-based studies have sought to confirm the association between atopic dermatitis, sleep and ADHD. In a German cross-sectional study, a significant association was observed between atopic dermatitis and ADHD (OR 1.54, p<0.001) with a stronger association seen in those with sleep problems (OR 2.67, p=0.001)124. More recently, analysis of cross-sectional data from 19 U.S.-based population surveys identified a pooled prevalence of atopic dermatitis of 10.1% among the 354,416 children included. A multivariate model that adjusted for a range of factors identified an association between atopic dermatitis and attention deficit disorder (ADD)/ADHD (Odds ratio 1.14; 95% CI 1.03–1.26). However, other risk factors were also identified that increased the risk of ADD/ADHD including disease severity and sleep disruption125.
Table 3: Association between atopic dermatitis severity, sleep and ADHD in the U.S. National Survey of Children’s Health 2007–2008 (adapted from Strom et al.125).
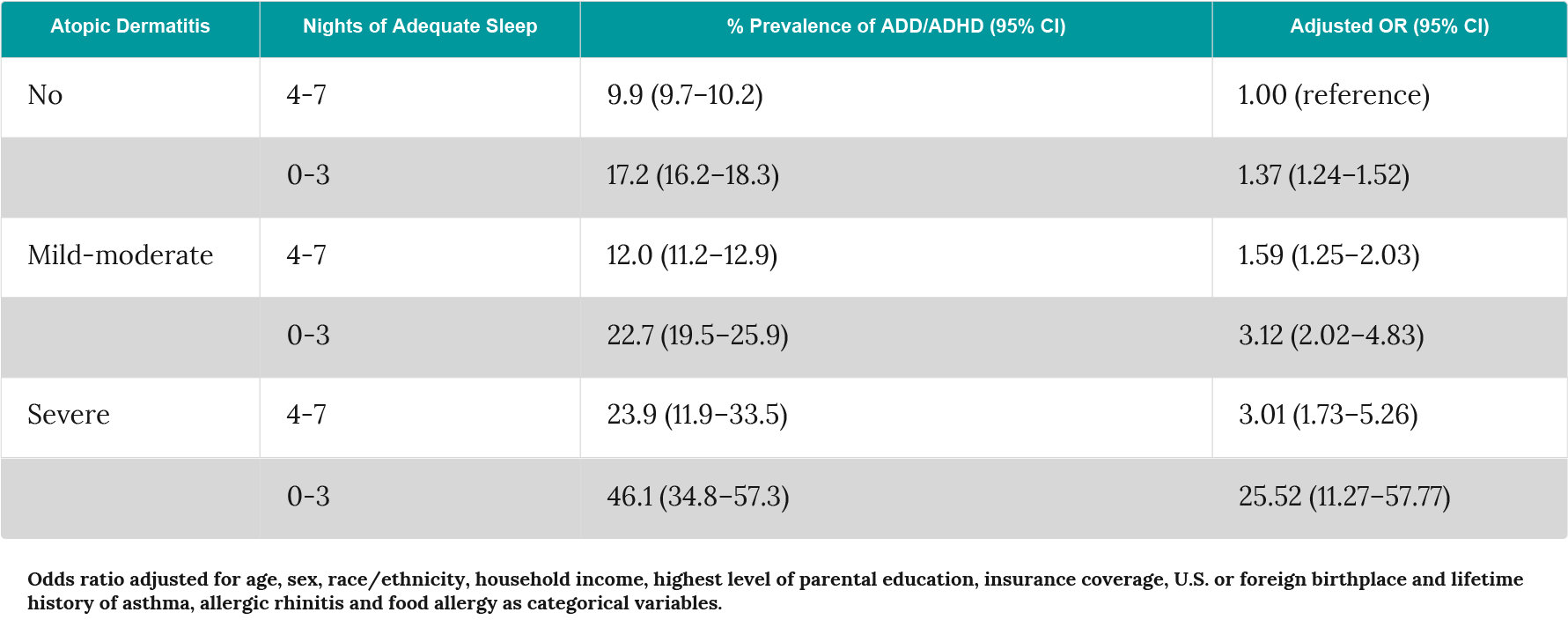
However, whether the presence of atopic dermatitis in pre-school children leads to increased levels of medication for ADHD remains uncertain. A Swedish birth cohort of 3,606 children found no association between pre-school (1–4 years of age) atopic dermatitis and prescription of ADHD treatments during school age (10–18 years of age) with a crude odds ratio of 1.16 (95% CI 0.83–1.61)126.
Mental health burden
For a number of years, an association has been established between atopic dermatitis and mental health issues with twice the rate of psychological disturbance in school-aged children with the condition versus controls127. Infants with atopic dermatitis have also been shown to be excessively clingy (50% vs. 10% in controls) and fearful (40% vs. 10% in controls)128. Recently, more well-defined mental health disorders have also been associated with atopic dermatitis including depression, anxiety, and autism. A cross-sectional analysis of the 2007 U.S. National Survey of Children’s Health found atopic dermatitis to be a significant risk factor for depression (OR 1.81; 95% CI 1.33–2.46), anxiety (OR 1.77; 95% CI 1.36–2.29), conduct disorder (OR 1.87; 95% CI 1.46–2.39) and autism (OR 3.04; 95% CI 2.13–4.34). The association also strengthened with increasing atopic dermatitis severity129.
The impact of atopic dermatitis on mental health continues into adolescence. A cross-sectional, questionnaire-based study of 3,775 adolescents (18–19 years) observed that those with atopic dermatitis were more likely to report mental health problems (OR 1.72; 95% CI 1.21–2.45), mental distress (OR 1.63; 95% CI 1.23–2.16) and, worryingly, suicidal ideation (15.5% vs. 9.1%; OR 1.87; 95% CI 1.31–2.68) compared to those without. In a subgroup analysis, those who had atopic dermatitis and itchy skin in the past week were significantly more likely to have these issues, with the odds ratio for suicidal ideation increasing substantially (OR 3.57; 95% CI 2.46–5.67)130.
Impact on physical and social activities
A survey of children in tertiary care for atopic dermatitis highlighted several physical and social functions that are impacted by the condition131.
Figure 11: Physical and social functions impacted by atopic dermatitis in children (adapted from Chamlin et al.131).
While the setting of this survey means the data is focussed on patients with more severe disease, subsequent studies have identified itching/scratching, impacts on sleep, treatment, sports and embarrassment related to the condition as those that most affect quality of life105. Embarrassment is a significant issue in patients with atopic dermatitis. In a survey of 429 patients, 45%, 54% and 77% of patients with mild, moderate and severe atopic dermatitis, respectively, reported feeling embarrassed or self-conscious in public sometimes, often or almost always. Levels of embarrassment are also linked to age with 90% of patients aged 8 to 15 years feeling embarrassed at least sometimes because of their condition132.
The psychologic, physical, and social impact of atopic dermatitis has a large impact on the quality of life of children and their families.
Restrictions placed on children with atopic dermatitis to help prevent and control their condition can have further implications, affecting children’s confidence, independence, and self-esteem. Such limitations, in addition to the chronic symptoms of atopic dermatitis, can also affect how they interact with their peers and how they perform at school133,134,135. Social isolation can be seen in children with atopic dermatitis from a young age. This may be adult admonitions regarding scratching for example, that can drive isolation or involve adults and children avoiding those with atopic dermatitis due to perceptions around contagion131,136. However, some parents also reported limiting interactions to avoid having to discuss their child’s skin131. In an international survey of 2,002 patients and caregivers, 39% of children (aged 8–17) in one study reported being teased or bullied while 24% (aged 2–13 years) and 36% (aged 14–17 years) said atopic dermatitis affects their self-confidence137. This has the potential to affect their relationships as children with severe atopic dermatitis had fewer friends, spent less leisure time with friends, and were less likely to be a member of a sports club138. However, social and leisure activities are affected with all disease severities with 31%, 55% and 86% of patients with mild, moderate and severe atopic dermatitis, respectively, experiencing ‘somewhat’, ‘a lot’ or ‘very much’ affected activities132. Furthermore, 30% (aged 2–13 years) and 46% (aged 14–17 years) of children with atopic dermatitis reported their school life to be affected and missed on average 2–3.5 school days per year due to their condition137. This continued into adolescence with 37% of patients at school not considered to be thriving130.
Family burden of atopic dermatitis
With atopic dermatitis placing such a significant burden on patients, frequently at an early age, it is unsurprising that family and carers also experience substantial burden. In fact, the social, personal and emotional impact of paediatric chronic atopic dermatitis on families was found to be greater than that of diabetes139.
Atopic dermatitis in children can affect their families/carers in several ways:
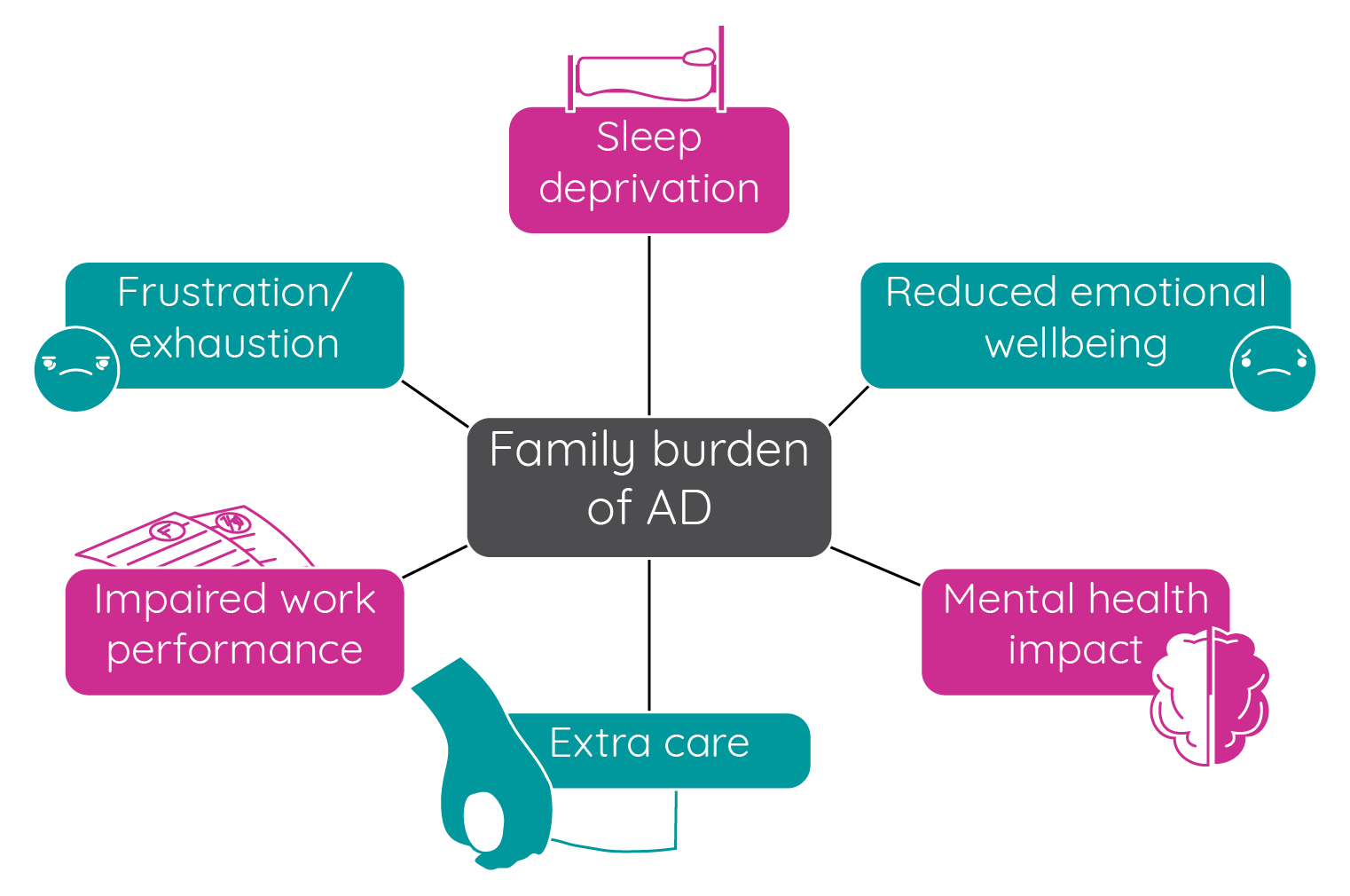
Figure 12: The impact of paediatric atopic dermatitis on family and carers (adapted from Blume-Peytavi et al.68; Drucker et al.105).
Caring for a child with atopic dermatitis can be time consuming. The day-to-day management of the condition can take a substantial amount of time, from around 1 hour140 to approximately 2–3 hours per day139, although conflicting results have been observed in other studies141. In addition, parents may also need to take time off work to look after their child142. Having a child with atopic dermatitis can also increase the amount of housework and preparation required, such as increased laundry, cleaning and food preparation with 90% of families in one study reporting problems with practical care. The impact of this can also lead to lifestyle limitations for the family and perhaps it is not surprising that the majority of parents report feelings of frustration, hopelessness, anxiety, depression, anger, guilt and the inability to cope143,131.
Having a child with atopic dermatitis also significantly impacts the sleep of family members. In a study of 270 patients and their parents, 61% of parents said their sleep was disturbed because of their child’s atopic dermatitis144. Meanwhile, a small study in the UK compared the sleep habits of 26 families with a child with atopic dermatitis and 29 families with a child with asthma. Interestingly, while the families of children with asthma spent no time at night caring for their child, the mothers of children with atopic dermatitis spent an average of 78 minutes per night attending to them while the fathers spent an average of 90 minutes145.
Burden of adult atopic dermatitis
While atopic dermatitis frequently resolves during childhood, it can continue into adulthood. A recent study suggested that approximately half of childhood cases extended into adult life146.
The burden of atopic dermatitis also continues into adulthood. A study of 349 adult patients with atopic dermatitis identified in the 2013 U.S. National Health and Wellness survey were compared with propensity score-matched patients without atopic dermatitis and patients with psoriasis. Compared to patients without atopic dermatitis, patients with atopic dermatitis had a significantly increased risk for arthritis (OR 1.7; 95% CI 1.3 – 2.3; p<0.001), asthma (OR 3.3; 95% CI 2.3–4.7; p<0.001) and nasal allergies/allergic rhinitis (OR 2.9; 95% CI 2.2–3.8; p<0.001). Surprisingly, the comorbidity risk was not significantly different between patients with mild atopic dermatitis and moderate-severe atopic dermatitis, although it was numerically higher in patients with moderate-severe disease147. When compared to patients with psoriasis, patients with atopic dermatitis had significantly higher odds of having comorbid asthma (OR 1.7; 95% CI 1.1–2.7; p=0.01), and nasal allergies/allergic rhinitis (OR 2.3; 95% CI 1.6–3.2; p<0.001) while patients with psoriasis had an increased risk of hypertension (OR 0.7; 95% CI 0.5–1.0; p=0.03)147. As in childhood atopic dermatitis, adults with atopic dermatitis also have significantly higher odds of ADD/ADHD (OR 1.61; 95% CI 1.25–2.06) after adjusting for sociodemographics, allergic disease and healthcare utilisation125.
Another article by Eckert et al. investigated the health-related quality of life (HRQoL) in the same patient population148.
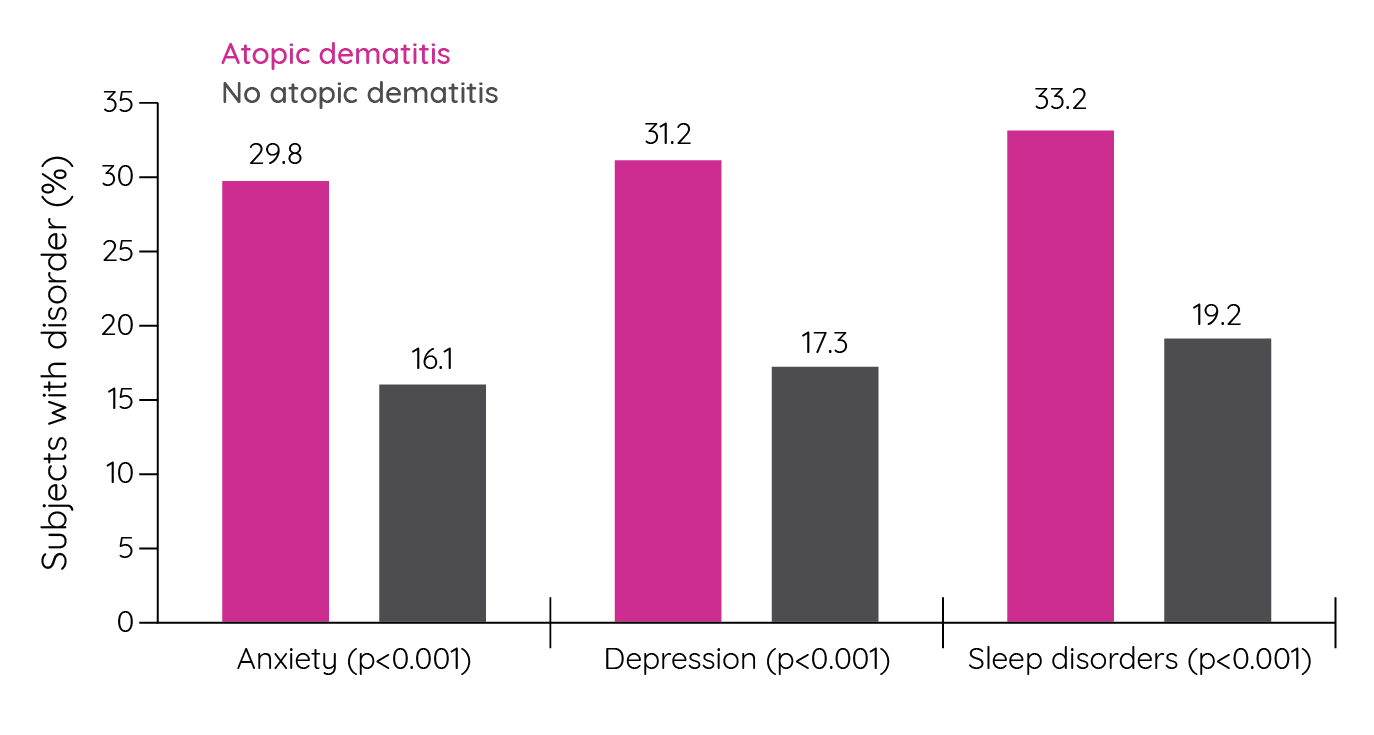
Figure 13: Percentage of patients with mood and sleep disorders (adapted from Eckert et al.148).
While significantly more patients with atopic dermatitis reported a mood or sleep disorder than patients without atopic dermatitis (Figure 13), no significant differences were seen when compared to patients with psoriasis. When comparing HRQoL, patients with atopic dermatitis had significantly worse mental and physical domains than patients without the condition, and comparable HRQoL to patients with psoriasis148. Similar results were seen in a cross-sectional survey of dermatological out-patients in 13 European countries. In this study, patients with dermatological conditions were significantly more likely to have depression than controls with atopic dermatitis (OR 3.27; 95% CI 1.61–6.62) and psoriasis (OR 3.02, 95% CI 1.86–4.90) having the third and fourth highest odds ratios, respectively behind leg ulcers and hand eczema149. Suicidal ideation was also seen to be significantly increased in adult patients with atopic dermatitis compared to healthy controls. In a cross-sectional survey of 181 patients and 64 controls, depressive symptoms, anxiety symptoms and suicidality were all significantly increased in patients with atopic dermatitis. In fact, 3.9% of patients, but no control subjects, presented with suicidality scores that might indicate an acute suicidal crisis150.
Work and productivity
The health implications of living with atopic dermatitis can also impact patients’ work productivity. In the 2010 U.S. National Health Interview Survey, 12.2% of participants with atopic dermatitis missed 1–2 days of work due to their condition while 2.3% reported missing 3 or more days20. Compared to people without atopic dermatitis, employed patients with atopic dermatitis reported higher absenteeism (9.9% vs. 3.6%; p<0.001), presenteeism (21.1% vs. 16.1%; p=0.027) and overall work impairment (25.6% vs. 18.1%; p=0.004). The impact of atopic dermatitis and psoriasis on productivity were comparable148. The burden was also comparable between patients with mild disease and moderate-severe disease. While the burden for absenteeism, presenteeism and overall work impairment was numerically higher in patients with more severe disease, it was not statistically significant148.
Interestingly, atopic dermatitis doesn’t appear to just affect people’s productivity at work but also their progression at work and even the work they do. While 14% of adults in the International Study on Life with Atopic Eczema believed that their career progression had been held back by their condition137, patients with atopic dermatitis have also reported avoiding careers where their skin may be an issue such as healthcare, food preparation, cleaning, hairdressing, and automobile repair151,152.
Financial burden of atopic dermatitis and resource utilisation
Calculating the financial costs of atopic dermatitis is challenging given how common and heterogeneous it is. The costs include prescriptions, physician visits, emergency and hospital costs and over-the-counter (OTC) pharmacy costs as well as indirect costs such as absenteeism and presenteeism153. These costs can be substantial, with 75% of patients reporting at least one doctor visit in the last year specifically for their atopic dermatitis20. More recently, comparison of patients with and without atopic dermatitis in the 2013 U.S. National Health and Wellness Survey revealed significant differences in healthcare resource use and costs147.
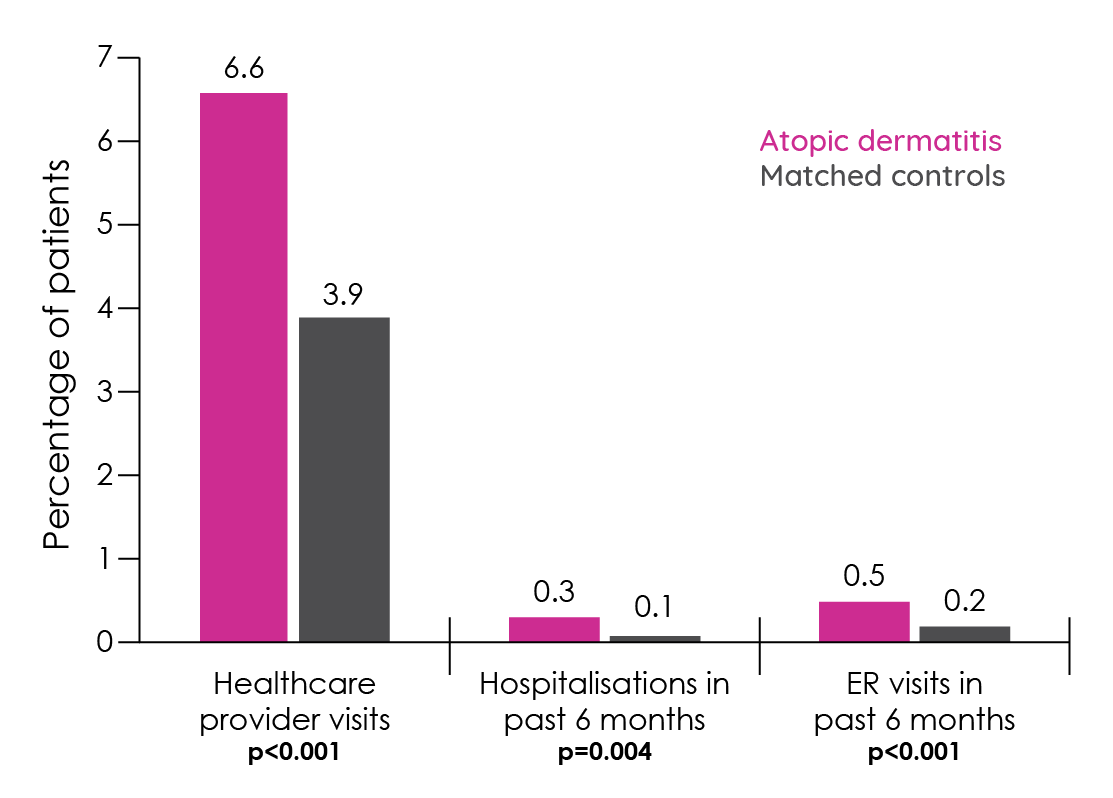
Figure 14: Healthcare resource utilisation in patients with atopic dermatitis versus matched controls without atopic dermatitis (adapted from Eckert et al.147).
A 2006 joint study between the American Academy of Dermatology and the Society for Investigative Dermatology sought to identify the direct and indirect costs for a range of skin diseases in the U.S. including atopic dermatitis. Direct costs of $1.009 billion and indirect costs of $3.219 billion gave a 2004 cost for managing atopic dermatitis of $4.228 billon154. This is the equivalent of well over $5 billion dollars today and may well be an underestimate of the true costs due to the use of low prevalence estimates, an absence of OTC product costs and some productivity losses153.
Diagnosing atopic dermatitis
Several classification criteria have been developed, but the Hanifin and Rajka criteria originally developed in 1980 remain the most widely used worldwide69.
Diagnosis of atopic dermatitis relies strongly on clinical presentation and is typically made through an extensive history and physical examination2.
The original criteria developed by Hanifin and Rajka are thorough, requiring 3 of 4 basic features (pruritus, typical morphology and distribution, chronic or chronically relapsing dermatitis, personal or family history of atopy) and at least 3 of 23 minor features to be met155. While comprehensive and validated, the extensive criteria make their application difficult in everyday clinical practice. Various revisions have been developed to aid using them in clinical practice including those developed by the UK Working Party who narrowed the criteria down to needing 1 mandatory and 5 major features to be met. These criteria have no requirement for laboratory testing and have been validated in various populations, however, they cannot be applied to very young children156,157,158,159.
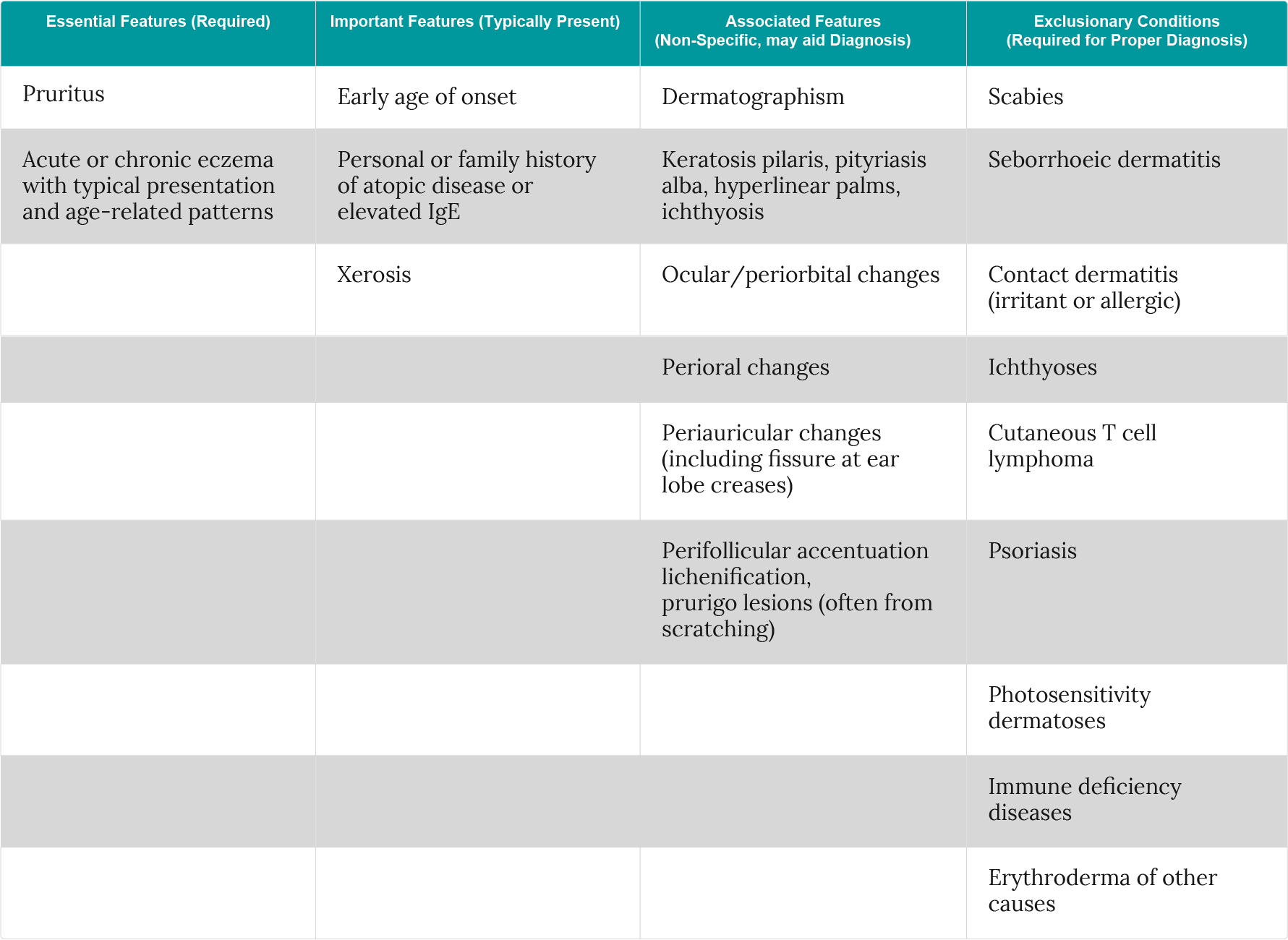
To address this limitation of the UK Working Group version, and to make the criteria more streamlined, the American Academy of Dermatology Consensus Conference proposed a further revision (Table 4)7. This version of the criteria has not been validated, but they were deemed suitable for diagnosing atopic dermatitis in patients of all ages2.
Table 4: American Academy of Dermatology consensus conference revision of the Hanifin & Rajka diagnostic criteria for atopic dermatitis (adapted from Eichenfield et al.7).
Watch Dr Amy Paller discuss how atopic dermatitis can affect patients and the impact this has on their lives
Biomarkers
At present, no laboratory biomarkers have been identified to aid with the diagnosis of atopic dermatitis. The elevation of total- or allergen specific IgE in serum or the use of skin tests to detect IgE-mediated sensitisation has been proposed. However, this feature is not present in all patients and has led to the terms ‘intrinsic’ (non-IgE-associated) and ‘extrinsic’ (IgE-associated) being used to describe these different forms of atopic dermatitis. However, controversy persists around the use of these terms and the practical applications of IgE detection in patients with atopic dermatitis69. Despite variation in IgE levels, it has been shown that intrinsic and extrinsic atopic dermatitis patient groups had similar underlying expression levels of Th2-related genes. This suggests that the underlying pathophysiology of their atopic dermatitis may be more similar than expected160.
Beyond diagnostic biomarkers, there is a general lack of biomarkers available to support patient stratification and management. In the past, treatment was typically nonspecific and consisted of topical and systemic immunosuppressants. However, many patients failed to respond to treatment leading to concerns among patients. With the advent of targeted biologic therapies in atopic dermatitis, identification of eligible patients who are likely to respond to therapy is now of increased need to reassure patients and avoid costly treatment failure161.
Numerous studies are currently ongoing looking at a variety of potential atopic dermatitis biomarkers. However, at present none have been shown to be reliable or sensitive enough to support regular clinical use2,69.
Measuring disease severity
Following a diagnosis of atopic dermatitis, it is necessary to assess disease severity. Given the presence of objective signs and subjective symptoms in most patients, a composite score is typically used to measure both aspects of the disease and the overall severity. The most commonly used score to achieve this is the Scoring of Atopic Dermatitis (SCORAD), which was developed in 1993162,69.
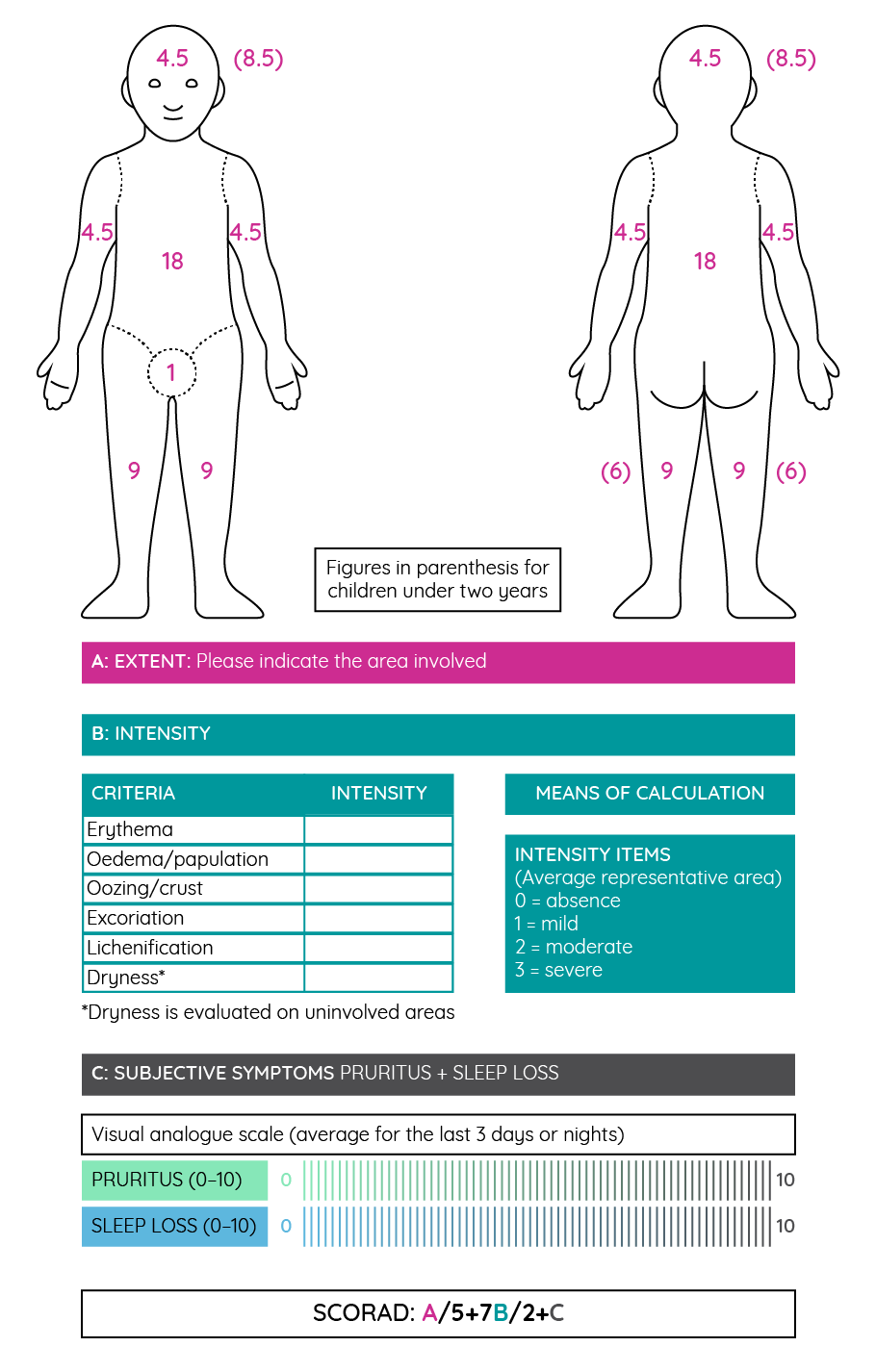
Figure 15: The Scoring of Atopic Dermatitis (SCORAD) tool for measuring disease severity (adapted from Stalder et al.162).
With a maximum possible SCORAD score of 103, patients with a score above 50 are typically considered to have severe atopic dermatitis, while those with a score below 25 are considered to have mild disease. Most cases of allergic dermatitis are mild with less than 10% considered severe. However, severe disease does seem to be more common in adult patients69. Treatment recommendations within guidelines are generally based on the severity of atopic dermatitis as measured by the SCORAD tool.
Dr Amy Paller details the impact of atopic dermatitis on patients with different levels of disease severity
Continue to the next section, focused on the treatment of atopic dermatitis, to find detailed of management goals for the condition, along with specific treatments available and in the pipeline.
References
- Nutten S. Atopic dermatitis: Global epidemiology and risk factors. Ann Nutr Metab. 2015;66:8–16.
- Avena-Woods C. Overview of atopic dermatitis. Am J Manag Care. 2017;23(8):S115–S123.
- Silverberg JI, Simpson EL. Association between severe eczema in children and multiple comorbid conditions and increased healthcare utilization. Pediatr Allergy Immunol. 2013;24(5):476–486.
- Silverberg JI, Simpson EL. Associations of childhood eczema severity: A US population-based study. Dermatitis. 2014;25(3):107–114.
- Emerson RM, Williams HC, Allen BR. Severity distribution of atopic dermatitis in the community and its relationship to secondary referral. Br J Dermatol. 1998;139(1):73–76.
- National Institute for Health and Care Excellence (NICE). Atopic eczema in under 12s: diagnosis and management. Clinical Guideline [CG57]. 2007. https://www.nice.org.uk/guidance/cg57. Accessed 18 June 2020.
- Eichenfield LF, Tom WL, Chamlin SL, Feldman SR, Hanifin JM, Simpson EL, et al. Guidelines of care for the management of atopic dermatitis: Section 1. Diagnosis and assessment of atopic dermatitis Work Group. J Am Acad Dermatol. 2014;70(2):338–351.
- Schneider L, Hanifin J, Boguniewicz M, Eichenfield LF, Spergel JM, Dakovic R, et al. Study of the atopic march: Development of atopic comorbidities. Pediatr Dermatol. 2016;33(4):388–398.
- Barnetson RSC, Rogers M. Childhood atopic eczema. Br Med J. 2002;324(7350):1376–1379.
- Margolis JS, Abuabara K, Bilker W, Hoffstad O, Margolis DJ. Persistence of mild to moderate atopic dermatitis. JAMA Dermatology. 2014;150(6):593–600.
- Kim JP, Chao LX, Simpson EL, Silverberg JI. Persistence of atopic dermatitis (AD): A systematic review and meta-analysis. J Am Acad Dermatol. 2016;75(4):681-687.e11.
- Margolis DJ, Mitra N. Heterogeneity of data included in meta-analysis on persistence of atopic dermatitis changes interpretation. J Am Acad Dermatol. 2017;76(5):e181.
- Silverberg JI, Simpson EL. Reply to: “Heterogeneity of data included in meta-analysis on persistence of atopic dermatitis alters interpretation”. J Am Acad Dermatol. 2017;76(5):e183–e184.
- Odhiambo JA, Williams HC, Clayton TO, Robertson CF, Asher MI, Aït-Khaled N, et al. Global variations in prevalence of eczema symptoms in children from ISAAC Phase Three. J Allergy Clin Immunol. 2009;124(6). doi:10.1016/j.jaci.2009.10.009.
- Williams H, Stewart A, von Mutius E, Cookson W, Anderson HR. Is eczema really on the increase worldwide? J Allergy Clin Immunol. 2008;121(4). doi:10.1016/j.jaci.2007.11.004.
- Silverberg JI. Public Health Burden and Epidemiology of Atopic Dermatitis. Dermatol Clin. 2017;35(3):283–289.
- Harrop J, Chinn S, Verlato G, Olivieri M, Norbäck D, Wjst M, et al. Eczema, atopy and allergen exposure in adults: A population-based study. Clin Exp Allergy. 2007;37(4):526–535.
- Hanifin JM, Reed ML, Drake LA, Koo J, Lebwohl MG, Leung DYM, et al. A population-based survey of eczema prevalence in the United States. Dermatitis. 2007;18(2):82–91.
- Silverberg JI, Hanifin JM. Adult eczema prevalence and associations with asthma and other health and demographic factors: A US population-based study. J Allergy Clin Immunol. 2013;132(5):1132–1138.
- Silverberg JI. Health care utilization, patient costs, and access to care in US adults with eczema: A population-based study. JAMA Dermatology. 2015;151(7):743–752.
- Barbarot S, Auziere S, Gadkari A, Girolomoni G, Puig L, Simpson EL, et al. Epidemiology of atopic dermatitis in adults: Results from an international survey. Allergy Eur J Allergy Clin Immunol. 2018;73(6):1284–1293.
- Kaufman BP, Guttman-Yassky E, Alexis AF. Atopic dermatitis in diverse racial and ethnic groups—Variations in epidemiology, genetics, clinical presentation and treatment. Exp Dermatol. 2018;27(4):340–357.
- Shaw TE, Currie GP, Koudelka CW, Simpson EL. Eczema prevalence in the United States: Data from the 2003 national survey of children’s health. J Invest Dermatol. 2011;131(1):67–73.
- Williams HC, Pembroke AC, Forsdyke H, Boodoo G, Hay RJ, Burney PGJ. London-born black caribbean children are at increased risk of atopic dermatitis. J Am Acad Dermatol. 1995;32(2 PART 1):212–217.
- McPherson T. Current understanding in pathogenesis of atopic dermatitis. Indian J Dermatol. 2016;61(6):649–655.
- Kim BE, Leung DYM. Significance of skin barrier dysfunction in atopic dermatitis. Allergy, Asthma Immunol Res. 2018;10(3):207–215.
- Cardona ID, Sang HC, Leung DYM. Role of bacterial superantigens in atopic dermatitis: Implications for future therapeutic strategies. Am J Clin Dermatol. 2006;7(5):273–279.
- Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, et al. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med. 2002;347(15):1151–1160.
- Merriman JA, Mueller EA, Cahill MP, Beck LA, Paller AS, Hanifin JM, et al. Temporal and Racial Differences Associated with Atopic Dermatitis Staphylococcus aureus and Encoded Virulence Factors. mSphere. 2016;1(6). doi:10.1128/msphere.00295-16.
- Simpson EL, Chalmers JR, Hanifin JM, Thomas KS, Cork MJ, McLean WHI, et al. Emollient enhancement of the skin barrier from birth offers effective atopic dermatitis prevention. J Allergy Clin Immunol. 2014;134(4):818–823.
- Glatz M, Jo JH, Kennedy EA, Polley EC, Segre JA, Simpson EL, et al. Emollient use alters skin barrier and microbes in infants at risk for developing atopic dermatitis. PLoS One. 2018;13(2). doi:10.1371/journal.pone.0192443.
- Sidbury R, Barton M. Advances in understanding and managing atopic dermatitis. F1000Research. 2015;4:1–7.
- Peng W, Novak N. Pathogenesis of atopic dermatitis. Clin Exp Allergy. 2015;45(3):566–574.
- Guttman-Yassky E, Hanifin JM, Boguniewicz M, Wollenberg A, Bissonnette R, Purohit V, et al. The role of phosphodiesterase 4 in the pathophysiology of atopic dermatitis and the perspective for its inhibition. Exp Dermatol. 2019;28(1):3–10.
- Scott IR, Harding CR. Filaggrin breakdown to water binding compounds during development of the rat stratum corneum is controlled by the water activity of the environment. Dev Biol. 1986;115(1):84–92.
- Denecker G, Hoste E, Gilbert B, Hochepied T, Ovaere P, Lippens S, et al. Caspase-14 protects against epidermal UVB photodamage and water loss. Nat Cell Biol. 2007;9(6):666–674.
- Nicotera P, Melino G. Caspase-14 and epidermis maturation. Nat Cell Biol. 2007;9(6):621–622.
- Jang H, Matsuda A, Jung K, Karasawa K, Matsuda K, Oida K, et al. Skin pH Is the master switch of kallikrein 5-mediated skin barrier destruction in a murine atopic dermatitis model. J Invest Dermatol. 2016;136(1):127–135.
- O’Regan GM, Sandilands A, McLean WHI, Irvine AD. Filaggrin in atopic dermatitis. J Allergy Clin Immunol. 2008;122(4):689–693.
- De Benedetto A, Rafaels NM, McGirt LY, Ivanov AI, Georas SN, Cheadle C, et al. Tight junction defects in patients with atopic dermatitis. J Allergy Clin Immunol. 2011;127(3):773-786.e7.
- Gruber R, Börnchen C, Rose K, Daubmann A, Volksdorf T, Wladykowski E, et al. Diverse Regulation of Claudin-1 and Claudin-4 in Atopic Dermatitis. Am J Pathol. 2015;185(10):2777–2789.
- Elias PM, Hatano Y, Williams ML. Basis for the barrier abnormality in atopic dermatitis: Outside-inside-outside pathogenic mechanisms. J Allergy Clin Immunol. 2008;121(6):1337–1343.
- Ali SM, Yosipovitch G. Skin pH: From basic science to basic skin care. Acta Derm Venereol. 2013;93(3):261–267.
- Leung DYM. New insights into atopic dermatitis: Role of skin barrier and immune dysregulation. Allergol Int. 2013;62(2):151–161.
- Danso M, Boiten W, van Drongelen V, Gmelig Meijling K, Gooris G, El Ghalbzouri A, et al. Altered expression of epidermal lipid bio-synthesis enzymes in atopic dermatitis skin is accompanied by changes in stratum corneum lipid composition. J Dermatol Sci. 2017;88(1):57–66.
- Ito S, Ishikawa J, Naoe A, Yoshida H, Hachiya A, Fujimura T, et al. Ceramide synthase 4 is highly expressed in involved skin of patients with atopic dermatitis. J Eur Acad Dermatology Venereol. 2017;31(1):135–141.
- Kim D, Lee NR, Park SY, Jun M, Lee K, Kim S, et al. As in Atopic Dermatitis, Nonlesional Skin in Allergic Contact Dermatitis Displays Abnormalities in Barrier Function and Ceramide Content. J Invest Dermatol. 2017;137(3):748–750.
- Li S, Villarreal M, Stewart S, Choi J, Ganguli-Indra G, Babineau DC, et al. Altered composition of epidermal lipids correlates with Staphylococcus aureus colonization status in atopic dermatitis. Br J Dermatol. 2017;177(4):e125–e127.
- Zimmermann M, Koreck A, Meyer N, Basinski T, Meiler F, Simone B, et al. TNF-like weak inducer of apoptosis (TWEAK) and TNF-α cooperate in the induction of keratinocyte apoptosis. J Allergy Clin Immunol. 2011;127(1). doi:10.1016/j.jaci.2010.11.005.
- Rebane A, Zimmermann M, Aab A, Baurecht H, Koreck A, Karelson M, et al. Mechanisms of IFN-γ-induced apoptosis of human skin keratinocytes in patients with atopic dermatitis. J Allergy Clin Immunol. 2012;129(5):1297–1306.
- Yoo J, Omori M, Gyarmati D, Zhou B, Aye T, Brewer A, et al. Spontaneous atopic dermatitis in mice expressing an inducible thymic stromal lymphopoietin transgene specifically in the skin. J Exp Med. 2005;202(4):541–549.
- Oyoshi MK, Larson RP, Ziegler SF, Geha RS. Mechanical injury polarizes skin dendritic cells to elicit a TH2 response by inducing cutaneous thymic stromal lymphopoietin expression. J Allergy Clin Immunol. 2010;126(5). doi:10.1016/j.jaci.2010.08.041.
- Novak N. An update on the role of human dendritic cells in patients with atopic dermatitis. J Allergy Clin Immunol. 2012;129(4):879–886.
- Boguniewicz M, Leung DYM. Atopic dermatitis: A disease of altered skin barrier and immune dysregulation. Immunol Rev. 2011;242(1):233–246.
- Bieber T. Atopic dermatitis. N Engl J Med. 2008;358(14):1483.
- Niebuhr M, Langnickel J, Draing C, Renz H, Kapp A, Werfel T. Dysregulation of toll-like receptor-2 (TLR-2)-induced effects in monocytes from patients with atopic dermatitis: Impact of the TLR-2 R753Q polymorphism. Allergy Eur J Allergy Clin Immunol. 2008;63(6):728–734.
- Niebuhr M, Lutat C, Sigel S, Werfel T. Impaired TLR-2 expression and TLR-2-mediated cytokine secretion in macrophages from patients with atopic dermatitis. Allergy Eur J Allergy Clin Immunol. 2009;64(11):1580–1587.
- Kuo IH, Carpenter-Mendini A, Yoshida T, McGirt LY, Ivanov AI, Barnes KC, et al. Activation of epidermal toll-like receptor 2 enhances tight junction function: Implications for atopic dermatitis and skin barrier repair. J Invest Dermatol. 2013;133(4):988–998.
- Otsuka A, Kabashima K. Mast cells and basophils in cutaneous immune responses. Allergy Eur J Allergy Clin Immunol. 2015;70(2):131–140.
- Nakamura Y, Oscherwitz J, Cease KB, Chan SM, Muñoz-Planillo R, Hasegawa M, et al. Staphylococcus δ-toxin induces allergic skin disease by activating mast cells. Nature. 2013;503(7476):397–401.
- Kusari A, Han AM, Schairer D, Eichenfield LF. Atopic Dermatitis: New Developments. Dermatol Clin. 2019;37(1):11–20.
- Cabanillas B, Brehler AC, Novak N. Atopic dermatitis phenotypes and the need for personalized medicine. Current Opinion in Allergy and Clinical Immunology. 2017;17(4):309–315.
- Schafer PH, Truzzi F, Parton A, Wu L, Kosek J, Zhang LH, et al. Phosphodiesterase 4 in inflammatory diseases: Effects of apremilast in psoriatic blood and in dermal myofibroblasts through the PDE4/CD271 complex. Cell Signal. 2016;28(7):753–763.
- Hanifin JM, Chan SC. Monocyte Phosphodiesterase Abnormalities and Dysregulation of Lymphoctye Function in Atopic Dermatitis. J Invest Dermatol. 1995;105(s1):84S-88S.
- Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 Receptor. Annu Rev Immunol. 2001;19(1):683–765.
- Baghoomian W, Na CH, Simpson EL. New and Emerging Biologics for Atopic Dermatitis. Am J Clin Dermatol. 2020. doi:10.1007/s40257-020-00515-1.
- Szalus K, Trzeciak M, Nowicki RJ. Jak-stat inhibitors in atopic dermatitis from pathogenesis to clinical trials results. Microorganisms. 2020;8(11):1–14.
- Blume-Peytavi U, Metz M. Atopic dermatitis in children: management of pruritus. J Eur Acad Dermatology Venereol. 2012;26:2–8.
- Wollenberg A, Barbarot S, Bieber T, Christen-Zaech S, Deleuran M, Fink-Wagner A, et al. Consensus-based European guidelines for treatment of atopic eczema (atopic dermatitis) in adults and children: part I. J Eur Acad Dermatology Venereol. 2018;32(5):657–682.
- Yosipovitch G, Goon ATJ, Wee J, Chan YH, Zucker I, Goh CL. Itch characteristics in Chinese patients with atopic dermatitis using a new questionnaire for the assessment of pruritus. Int J Dermatol. 2002;41(4):212–216.
- Dawn A, Papoiu ADP, Chan YH, Rapp SR, Rassette N, Yosipovitch G. Itch characteristics in atopic dermatitis: Results of a web-based questionnaire. Br J Dermatol. 2009;160(3):642–644.
- Umehara Y, Kiatsurayanon C, Trujillo-Paez JV, Chieosilapatham P, Peng G, Yue H, et al. Intractable Itch in Atopic Dermatitis: Causes and Treatments. Biomedicines. 2021;9(3):229.
- Mollanazar NK, Smith PK, Yosipovitch G. Mediators of Chronic Pruritus in Atopic Dermatitis: Getting the Itch Out? Clin Rev Allergy Immunol. 2016;51(3):263–292.
- Lodén M. Effect of moisturizers on epidermal barrier function. Clin Dermatol. 2012;30(3):286–296.
- Ny A, Egelrud T. Epidermal Hyperproliferation and Decreased Skin Barrier Function in Mice Overexpressing Stratum Corneum Chymotryptic Enzyme. Acta Derm Venereol. 2004;84(1):18–22.
- Lee CH, Chuang HY, Shih CC, Jong SB, Chang CH, Yu HS. Transepidermal water loss, serum IgE and β-endorphin as important and independent biological markers for development of itch intensity in atopic dermatitis. Br J Dermatol. 2006;154(6):1100–1107.
- Wahlgren CF, Hägermark Ö, Linder MT, Scheynius A. Itch and inflammation induced by intradermally injected interleukin-2 in atopic dermatitis patients and healthy subjects. Arch Dermatol Res. 1995;287(6):572–580.
- Gaspari AA, Lotze MT, Rosenberg SA, Stern JB, Katz SI. Dermatologic Changes Associated With Interleukin 2 Administration. JAMA J Am Med Assoc. 1987;258(12):1624–1629.
- Lee RE, Gaspari AA, Lotze MT, Chang AE, Rosenberg SA. Interleukin 2 and Psoriasis. Arch Dermatol. 1988;124(12):1811–1815.
- Kremer AE, Feramisco J, Reeh PW, Beuers U, Oude Elferink RPJ. Receptors, cells and circuits involved in pruritus of systemic disorders. Biochim Biophys Acta - Mol Basis Dis. 2014;1842(7):869–892.
- Wahlgren CF, Scheynius A, Hagermark O. Antipruritic effect of oral cyclosporin A in atopic dermatitis. Acta Derm Venereol. 1990;70(4):323–329.
- Jeong CW, Ahnt KS, Rho NK, Park YD, Lee DY, Lee JH, et al. Differential in vivo cytokine mRNA expression in lesional skin of intrinsic vs. extrinsic atopic dermatitis patients using semiquantitative RT-PCR. Clin Exp Allergy. 2003;33(12):1717–1724.
- Metwally SS, Mosaad YM, Abdel-Samee ER, El-Gayyar MA, Abdel-Aziz AM, El-Chennawi FA. IL-13 gene expression in patients with atopic dermatitis: relation to IgE level and to disease severity. Egypt J Immunol. 2004;11(2):171–177.
- Beck LA, Thaçi D, Hamilton JD, Graham NM, Bieber T, Rocklin R, et al. Dupilumab Treatment in Adults with Moderate-to-Severe Atopic Dermatitis. N Engl J Med. 2014;371(2):130–139.
- Oldhoff JM, Darsow U, Werfel T, Katzer K, Wulf A, Laifaoui J, et al. Anti-IL-5 recombinant humanized monoclonal antibody (Mepolizumab) for the treatment of atopic dermatitis. Allergy Eur J Allergy Clin Immunol. 2005;60(5):693–696.
- Paller AS, Kabashima K, Bieber T. Therapeutic pipeline for atopic dermatitis: End of the drought? J Allergy Clin Immunol. 2017;140(3):633–643.
- Sonkoly E, Muller A, Lauerma AI, Pivarcsi A, Soto H, Kemeny L, et al. IL-31: A new link between T cells and pruritus in atopic skin inflammation. J Allergy Clin Immunol. 2006;117(2):411–417.
- Raap U, Wichmann K, Bruder M, Ständer S, Wedi B, Kapp A, et al. Correlation of IL-31 serum levels with severity of atopic dermatitis. J Allergy Clin Immunol. 2008;122(2):421–423.
- Kasraie S, Niebuhr M, Werfel T. Interleukin (IL)-31 activates signal transducer and activator of transcription (STAT)-1, STAT-5 and extracellular signal-regulated kinase 1/2 and down-regulates IL-12p40 production in activated human macrophages. Allergy Eur J Allergy Clin Immunol. 2013;68(6):739–747.
- Hawro T, Saluja R, Weller K, Altrichter S, Metz M, Maurer M. Interleukin-31 does not induce immediate itch in atopic dermatitis patients and healthy controls after skin challenge. Allergy Eur J Allergy Clin Immunol. 2014;69(1):113–117.
- Kroeger KM, Sullivan BM, Locksley RM. IL-18 and IL-33 elicit Th2 cytokines from basophils via a MyD88- and p38α-dependent pathway. J Leukoc Biol. 2009;86(4):769–778.
- Rankin AL, Mumm JB, Murphy E, Turner S, Yu N, McClanahan TK, et al. IL-33 Induces IL-13–Dependent Cutaneous Fibrosis. J Immunol. 2010;184(3):1526–1535.
- Savinko T, Matikainen S, Saarialho-Kere U, Lehto M, Wang G, Lehtimäki S, et al. IL-33 and ST2 in atopic dermatitis: Expression profiles and modulation by triggering factors. J Invest Dermatol. 2012;132(5):1392–1400.
- Tamagawa-Mineoka R, Okuzawa Y, Masuda K, Katoh N. Increased serum levels of interleukin 33 in patients with atopic dermatitis. J Am Acad Dermatol. 2014;70(5):882–888.
- Petra AI, Tsilioni I, Taracanova A, Katsarou-Katsari A, Theoharides TC. Interleukin 33 and interleukin 4 regulate interleukin 31 gene expression and secretion from human laboratory of allergic diseases 2 mast cells stimulated by substance P and/or immunoglobulin e. Allergy Asthma Proc. 2018;39(2):153–160.
- Moniaga CS, Jeong SK, Egawa G, Nakajima S, Hara-Chikuma M, Jeon JE, et al. Protease activity enhances production of thymic stromal lymphopoietin and basophil accumulation in flaky tail mice. Am J Pathol. 2013;182(3):841–851.
- Wilson SR, Thé L, Batia LM, Beattie K, Katibah GE, McClain SP, et al. The epithelial cell-derived atopic dermatitis cytokine TSLP activates neurons to induce itch. Cell. 2013;155(2):285–295.
- Ziegler SF, Roan F, Bell BD, Stoklasek TA, Kitajima M, Han H. The Biology of Thymic Stromal Lymphopoietin (TSLP). Adv Pharmacol. 2013;66:129–155.
- Jariwala SP, Abrams E, Benson A, Fodeman J, Zheng T. The role of thymic stromal lymphopoietin in the immunopathogenesis of atopic dermatitis. Clin Exp Allergy. 2011;41(11):1515–1520.
- Rinaldi G. The Itch-Scratch Cycle: A Review of the Mechanisms. Dermatol Pract Concept. 2019;9(2):90–97.
- Johanek LM, Meyer RA, Hartke T, Hobelmann JG, Maine DN, LaMotte RH, et al. Psychophysical and physiological evidence for parallel afferent pathways mediating the sensation of itch. J Neurosci. 2007;27(28):7490–7497.
- Namer B, Carr R, Johanek LM, Schmelz M, Handwerker HO, Ringkamp M. Separate peripheral pathways for pruritus in man. J Neurophysiol. 2008;100(4):2062–2069.
- Alexander JOD. The physiology of itch. Parasitology Today. 1986;2(12):345–351.
- Zhao ZQ, Liu XY, Jeffry J, Karunarathne WKA, Li JL, Munanairi A, et al. Descending control of itch transmission by the serotonergic system via 5-HT1A-facilitated GRP-GRPR signaling. Neuron. 2014;84(4):821–834.
- Drucker AM, Wang AR, Li WQ, Sevetson E, Block JK, Qureshi AA. The Burden of Atopic Dermatitis: Summary of a Report for the National Eczema Association. J Invest Dermatol. 2017;137(1):26–30.
- Silverberg JI, Vakharia PP, Chopra R, Sacotte R, Patel N, Immaneni S, et al. Phenotypical Differences of Childhood- and Adult-Onset Atopic Dermatitis. J Allergy Clin Immunol Pract. 2018;6(4):1306–1312.
- Karimkhani C, Dellavalle RP, Coffeng LE, Flohr C, Hay RJ, Langan SM, et al. Global skin disease morbidity and mortality an update from the global burden of disease study 2013. JAMA Dermatology. 2017;153(5):406–412.
- Beattie PE, Lewis-Jones MS. A comparative study of impairment of quality of life in children with skin disease and children with other chronic childhood diseases. Br J Dermatol. 2006;155(1):145–151.
- Schmitt J, Csötönyi F, Bauer A, Meurer M. Determinants of treatment goals and satisfaction of patients with atopic eczema. JDDG - J Ger Soc Dermatology. 2008;6(6):458–465.
- Vakharia PP, Chopra R, Sacotte R, Patel KR, Singam V, Patel N, et al. Burden of skin pain in atopic dermatitis. Ann Allergy, Asthma Immunol. 2017;119(6):548-552.e3.
- Gustafsson D, Sjöberg O, Foucard T. Development of allergies and asthma in infants and young children with atopic dermatitis - A prospective follow-up to 7 years age. Allergy Eur J Allergy Clin Immunol. 2000;55(3):240–245.
- van der Hulst AE, Klip H, Brand PLP. Risk of developing asthma in young children with atopic eczema: A systematic review. J Allergy Clin Immunol. 2007;120(3):565–569.
- Martin PE, Matheson MC, Gurrin L, Burgess JA, Osborne N, Lowe AJ, et al. Childhood eczema and rhinitis predict atopic but not nonatopic adult asthma: A prospective cohort study over 4 decades. J Allergy Clin Immunol. 2011;127(6). doi:10.1016/j.jaci.2011.02.041.
- Ellinghaus D, Baurecht H, Esparza-Gordillo J, Rodríguez E, Matanovic A, Marenholz I, et al. High-density genotyping study identifies four new susceptibility loci for atopic dermatitis. Nat Genet. 2013;45(7):808–812.
- Schmitt J, Schwarz K, Baurecht H, Hotze M, Fölster-Holst R, Rodríguez E, et al. Atopic dermatitis is associated with an increased risk for rheumatoid arthritis and inflammatory bowel disease, and a decreased risk for type 1 diabetes. J Allergy Clin Immunol. 2016;137(1):130–136.
- Hart CN, Palermo TM, Rosen CL. Health-related quality of life among children presenting to a pediatric sleep disorders clinic. Behav Sleep Med. 2005;3(1):4–17.
- Hiscock H, Bayer J, Gold L, Hampton A, Ukoumunne OC, Wake M. Improving infant sleep and maternal mental health: A cluster randomised trial. Arch Dis Child. 2007;92(11):952–958.
- Gregory AM, Van Der Ende J, Willis TA, Verhulst FC. Parent-reported sleep problems during development and self-reported anxiety/depression, attention problems, and aggressive behavior later in life. Arch Pediatr Adolesc Med. 2008;162(4):330–335.
- Yokomaku A, Misao K, Omoto F, Yamagishi R, Tanaka K, Takada K, et al. A study of the association between sleep habits and problematic behaviors in preschool children. Chronobiol Int. 2008;25(4):549–564.
- Reid P, Lewis-Jones MS. Sleep difficulties and their management in preschoolers with atopic eczema. Clin Exp Dermatol. 1995;20(1):38–41.
- Wittkowski A, Richards HL, Griffiths CEM, Main CJ. Illness perception in individuals with atopic dermatitis. Psychol Heal Med. 2007;12(4):433–444.
- Camfferman D, Kennedy JD, Gold M, Martin AJ, Lushington K. Eczema and sleep and its relationship to daytime functioning in children. Sleep Med Rev. 2010;14(6):359–369.
- Drucker AM. Atopic dermatitis: Burden of illness, quality of life, and associated complications. Allergy Asthma Proc. 2017;38(1):3–8.
- Romanos M, Gerlach M, Warnke A, Schmitt J. Association of attention-deficit/hyperactivity disorder and atopic eczema modified by sleep disturbance in a large population-based sample. J Epidemiol Community Health. 2010;64(3):269–273.
- Strom MA, Fishbein AB, Paller AS, Silverberg JI. Association between atopic dermatitis and attention deficit hyperactivity disorder in U.S. children and adults. Br J Dermatol. 2016;175(5):920–929.
- Johansson EK, Ballardini N, Kull I, Bergström A, Wahlgren CF. Association between preschool eczema and medication for attention-deficit/hyperactivity disorder in school age. Pediatr Allergy Immunol. 2017;28(1):44–50.
- Absolon CM, Cottrell D, Eldridge SM, Glover MT. Psychological disturbance in atopic eczema: the extent of the problem in school‐aged children. Br J Dermatol. 1997;137(2):241–245.
- Daud LR, Garralda ME, David TJ. Psychosocial adjustment in preschool children with atopic eczema. Arch Dis Child. 1993;69(6):670–676.
- Yaghmaie P, Koudelka CW, Simpson EL. Mental health comorbidity in patients with atopic dermatitis. J Allergy Clin Immunol. 2013;131(2):428–433.
- Halvorsen JA, Lien L, Dalgard F, Bjertness E, Stern RS. Suicidal ideation, mental health problems, and social function in adolescents with eczema: A population-based study. J Invest Dermatol. 2014;134(7):1847–1854.
- Chamlin SL, Frieden IJ, Williams ML, Chren MM. Effects of atopic dermatitis on young American children and their families. Pediatrics. 2004;114(3):607–611.
- Paller AS, Mcalister RO, Doyle JJ, Jackson A. Perceptions of physicians and pediatric patients about atopic dermatitis, its impact, and its treatment. Clin Pediatr (Phila). 2002;41(5):323–332.
- Ward S. The Effective Management of Atopic Dermatitis in School-Age Children. Nurs Times. 2004;100(32):55–56.
- Lee S-I, Kim J, Han Y, Ahn K. A proposal: Atopic Dermatitis Organizer (ADO) guideline for children. Asia Pac Allergy. 2011;1(2):53.
- Schmitt J, Chen CM, Apfelbacher C, Romanos M, Lehmann I, Herbarth O, et al. Infant eczema, infant sleeping problems, and mental health at 10 years of age: The prospective birth cohort study LISAplus. Allergy Eur J Allergy Clin Immunol. 2011;66(3):404–411.
- Chernyshov P V. Stigmatization and self-perception in children with atopic dermatitis. Clin Cosmet Investig Dermatol. 2016;9:159–166.
- Zuberbier T, Orlow SJ, Paller AS, Taïeb A, Allen R, Hernanz-Hermosa JM, et al. Patient perspectives on the management of atopic dermatitis. J Allergy Clin Immunol. 2006;118(1):226–232.
- Brenninkmeijer EEA, Legierse CM, Sillevis Smitt JH, Last BF, Grootenhuis MA, Bos JD. The course of life of patients with childhood atopic dermatitis. Pediatr Dermatol. 2009;26(1):14–22.
- Su JC, Kemp AS, Varigos GA, Nolan TM. Atopic eczema: Its impact on the family and financial cost. Arch Dis Child. 1997;76(2):159–162.
- Holm EA, Jemec GBE. Time spent on treatment of atopic dermatitis: A new method of measuring pediatric morbidity? Pediatr Dermatol. 2004;21(6):623–627.
- Jemec GB, Esmann S, Holm E, Tycho A, Jørgensen TM. Time spent on treatment (TSOT). An independent assessment of disease severity in atopic dermatitis. Acta Dermatovenerol Alp Pannonica Adriat. 2006;15(3):119–24.
- Lewis-Jones S. Quality of life and childhood atopic dermatitis: The misery of living with childhood eczema. Int J Clin Pract. 2006;60(8):984–992.
- Lawson V, Lewis-Jones MS, Finlay AY, Reid P, Owens RG. The family impact of childhood atopic dermatitis: The Dermatitis Family Impact questionnaire. Br J Dermatol. 1998;138(1):107–113.
- Chamlin SL, Mattson CL, Frieden IJ, Williams ML, Mancini AJ, Cella D, et al. The price of pruritus: Sleep disturbance and cosleeping in atopic dermatitis. Arch Pediatr Adolesc Med. 2005;159(8):745–750.
- Moore K, David TJ, Murray CS, Child F, Arkwright PD. Effect of childhood eczema and asthma on parental sleep and well-being: A prospective comparative study. Br J Dermatol. 2006;154(3):514–518.
- Napolitano M, Megna M, Patruno C, Gisondi P, Ayala F, Balato N. Adult atopic dermatitis: a review. G Ital Dermatol Venereol. 2016;151(4):403–11.
- Eckert L, Gupta S, Amand C, Gadkari A, Mahajan P, Gelfand JM. The burden of atopic dermatitis in US adults: Health care resource utilization data from the 2013 National Health and Wellness Survey. J Am Acad Dermatol. 2018;78(1):54-61.e1.
- Eckert L, Gupta S, Amand C, Gadkari A, Mahajan P, Gelfand JM. Impact of atopic dermatitis on health-related quality of life and productivity in adults in the United States: An analysis using the National Health and Wellness Survey. J Am Acad Dermatol. 2017;77(2):274-279.e3.
- Dalgard FJ, Gieler U, Tomas-Aragones L, Lien L, Poot F, Jemec GBE, et al. The Psychological Burden of Skin Diseases: A Cross-Sectional Multicenter Study among Dermatological Out-Patients in 13 European Countries. J Invest Dermatol. 2015;135(4):984–991.
- Dieris-Hirche J, Gieler U, Petrak F, Milch W, te Wildt B, Dieris B, et al. Suicidal ideation in adult patients with atopic dermatitis: A German cross-sectional study. Acta Derm Venereol. 2017;97(10):1189–1195.
- Nyrén M, Lindberg M, Stenberg B, Svensson M, Svensson Å, Meding B. Influence of childhood atopic dermatitis on future worklife. Scand J Work Environ Heal. 2005;31(6):474–478.
- Holm EA, Esmann S, Jemec GBE. The handicap caused by atopic dermatitis - Sick leave and job avoidance. J Eur Acad Dermatology Venereol. 2006;20(3):255–259.
- Drucker AM, Qureshi AA, Amand C, Villeneuve S, Gadkari A, Chao J, et al. Health Care Resource Utilization and Costs Among Adults with Atopic Dermatitis in the United States: A Claims-Based Analysis. J Allergy Clin Immunol Pract. 2018;6(4):1342–1348.
- Bickers DR, Lim HW, Margolis D, Weinstock MA, Goodman C, Faulkner E, et al. The burden of skin diseases: 2004. A joint project of the American Academy of Dermatology Association and the Society for Investigative Dermatology. J Am Acad Dermatol. 2006;55(3):490–500.
- Hanifin JM, Rajka G. Diagnostic features of atopic dermatitis. Acta Derm Venereol. 1980;92:44–47.
- Williams HC, Burney PG, Hay RJ, Archer CB, Shipley MJ, Hunter JJ, et al. The U.K. Working Party’s Diagnostic Criteria for Atopic Dermatitis. I. Derivation of a minimum set of discriminators for atopic dermatitis. Br J Dermatol. 1994;131(3):383–396.
- Williams HC, Burney PG, Pembroke AC, Hay RJ. Validation of the U.K. Diagnostic Criteria for Atopic Dermatitis in a Population Setting. U.K. Diagnostic Criteria for Atopic Dermatitis Working Party. Br J Dermatol. 1996;135(1):12–7.
- Gu H, Chen XS, Chen K, Yan Y, Jing H, Chen XQ, et al. Evaluation of diagnostic criteria for atopic dermatitis: Validity of the criteria of Williams et al. in a hospital-based setting. Br J Dermatol. 2001;145(3):428–433.
- De D, Kanwar AJ, Handa S. Comparative efficacy of Hanifin and Rajka’s criteria and the UK working party’s diagnostic criteria in diagnosis of atopic dermatitis in a hospital setting in North India. J Eur Acad Dermatology Venereol. 2006;20(7):853–859.
- Suárez-Fariñas M, Dhingra N, Gittler J, Shemer A, Cardinale I, De Guzman Strong C, et al. Intrinsic atopic dermatitis shows similar TH2 and higher T H17 immune activation compared with extrinsic atopic dermatitis. J Allergy Clin Immunol. 2013;132(2):361–370.
- Thijs JL, de Bruin-Weller MS, Hijnen DJ. Current and Future Biomarkers in Atopic Dermatitis. Immunol Allergy Clin North Am. 2017;37(1):51–61.
- Stalder JF, Taïeb A, Atherton DJ, Bieber P, Bonifazi E, Broberg A, et al. Severity scoring of atopic dermatitis: The SCORAD index: Consensus report of the european task force on atopic dermatitis. Dermatology. 1993;186(1):23–31.
of interest
are looking at
saved
next event
This content has been developed by Medthority who previously received funding in order to help provide its healthcare professional members with access to the highest quality medical and scientific information, education and associated relevant content.