
Atopic Dermatitis treatment
Atopic dermatitis is a common inflammatory skin disease predominantly affecting children1. Learn about the current and upcoming treatment classes available to manage the condition and prevent acute flares.
- Gain clarity regarding classical and targeted treatment options available to alleviate symptoms and prevent an atopic march
- Look to current research and future possibility for treatments which can target specific pathways and inflammatory mediators
- Understand the treatment algorithm, including when and how to use specific therapies for varying severities of atopic dermatitis
Treatment goals for atopic dermatitis
While atopic dermatitis may spontaneously resolve in patients, it is not curable. Many patients will experience chronic disease and so the aims of treatment are limited to2:
- Minimise the number of flares
- Reduce the duration and degree of any flares
Interestingly, when 3,846 dermatology patients across 13 countries were asked whether they had treatment-related issues, 63.4% of patients with atopic dermatitis said they did – more than any other skin disease assessed3.
80% of patients in an online survey were dissatisfied with their atopic dermatitis treatment4
The topical treatments for atopic dermatitis have changed little over the past 15 years, consisting of topical corticosteroids (TCS) and topical calcineurin inhibitors (TCI). The effectiveness of TCS has been demonstrated over the years, however, local and systemic adverse effects limit their use. This is of particular concern in children where a greater surface area to body weight ratio increases the potential for elevated exposure5.
Professor Michael Cork explains a significant unmet need in atopic dermatitis and considers the potential limitations of treatment in relation to the need for additional therapeutics to become available.
Table 1: Adverse effects associated with the use of topical corticosteroids (adapted from Zane et al.5).
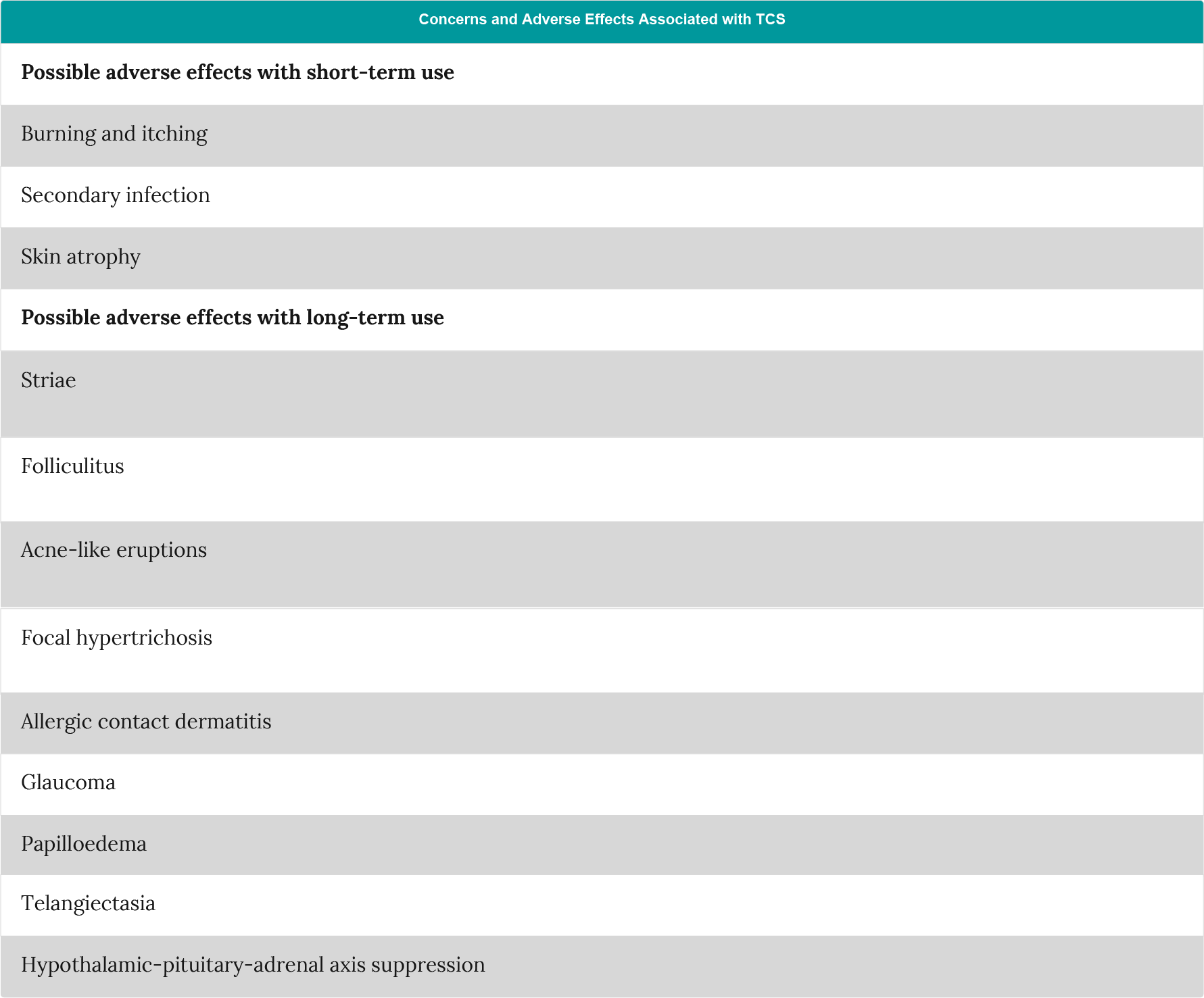
While many patients are satisfied with the efficacy of TCS treatment, concerns over their use are common. Steroid phobia, or corticophobia, has been observed in over 80% of patients and parents6,7, impacting treatment compliance and failure7. Interestingly, a study of steroid phobia among French dermatologists, paediatricians and pharmacists identified concerns among all groups, but particularly among pharmacists, which may lead to increased concerns among patients8. A separate study also observed concern among French pharmacists around the use of topical corticosteroids in patients with atopic dermatitis, with this lack of trust potentially helping to maintain the fear of corticosteroids among parents and patients9. A validated questionnaire (TOPICOP©) has now been developed enabling patients’ topical corticosteroid phobia to be assessed in everyday clinical practice, helping to identify patients requiring additional support and education10,11.
Two TCIs are currently available for patients who are inadequately responsive to, or intolerant of, TCS. However, TCIs are also associated with application site burning and stinging as well as a small increase in the incidence of herpes simplex infection12,5 although no increased risk of infection was observed versus TCS treatment in a 5-year study of pimecrolimus in mild-to-moderate atopic dermatitis13. Of greater concern has been the historical link between TCI and increased lymphoma risk. In 2006, a boxed warning was added to the tacrolimus and pimecrolimus labels. Recent epidemiologic data suggests that the incidence of lymphoma was no greater in TCI-treated patients than the general population. However, U.S. FDA deemed an association may still exist and kept the boxed warning in place5.
Patient satisfaction with treatment
There remains significant unmet need for new therapeutic treatments with improved efficacy and/or safety profiles. Just 46% of 961 patients reported their prescription medications as ‘'moderately’ or ‘very’ effective in one survey14. This perception of treatment inefficacy can translate into treatment dissatisfaction.
Patients ‘not at all’ or ‘fairly’ satisfied with current treatments15
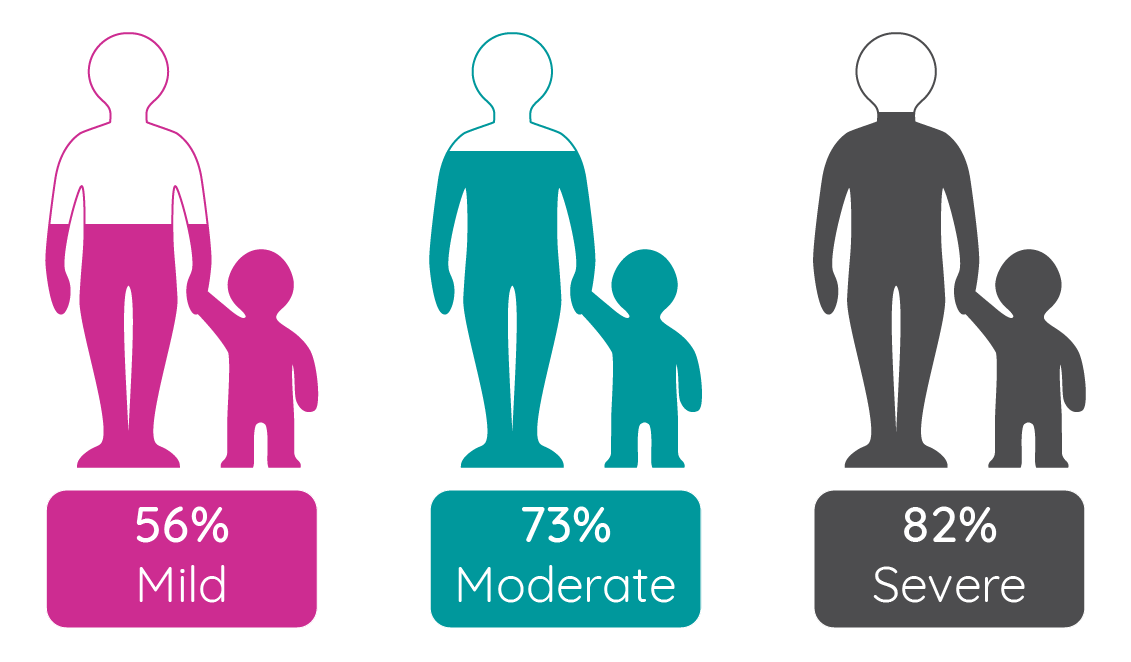
Figure 1: Dissatisfaction with current atopic dermatitis treatments (adapted from Paller et al.15). Dissatisfaction was recorded for a “not-at-all” or “fairly” satisfied patient response.
A Japanese study of 1,327 patients reported treatment dissatisfaction in 43.6% of patients16 while a cross-sectional study of 226 patients using a visual analogue scale score to assess treatment satisfaction (0, very dissatisfied; 100, very satisfied) showed an average score of 59.6 (±30.8) suggesting moderate satisfaction. However, the broad range of scores suggests substantial variability in the levels of patient satisfaction. It is worth noting that disease severity explained only 20% of the variance seen in treatment satisfaction. The most important determinant of treatment satisfaction was actually the perceived competence of the treating physician17.
Such concerns around treatment effectiveness do not seem to be shared by many physicians reflecting a disconnect in patient–physician communication.
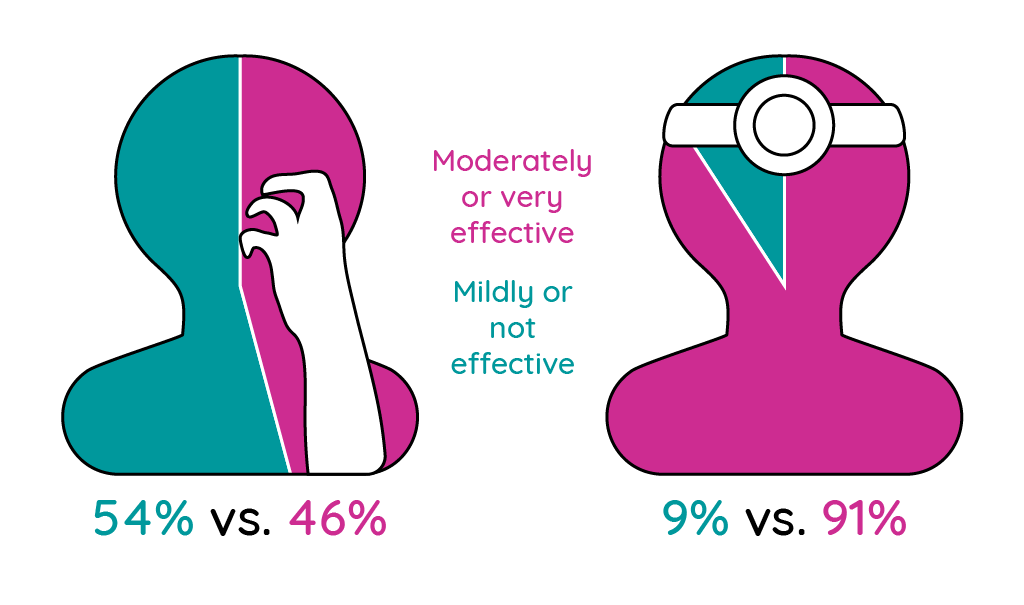
Figure 2: Prescription medications for atopic dermatitis regarded as moderately or very effective by patients and physicians (adapted from McAlister et al.14; Paller et al.15).
Interestingly, 47% of physicians thought that their patients were not satisfied with their treatment citing a lack of efficacy as the main cause of dissatisfaction15.
Discordance between patient and physician opinions on treatment are also apparent with different formulations. While nearly two-thirds of physicians preferred ointment products, most physicians believed that patients overwhelmingly preferred creams. However, when patients were asked, there was a near even divide between ointments and creams with older children preferring ointments15.
Watch Dr Amy Paller’s expert opinion on identifying the primary treatment goals of atopic dermatitis
Current treatment options offer mixed efficacy and significant adverse events. It is apparent from patient satisfaction data that additional therapies are needed to meet this unmet need. Furthermore, some of the concerns surrounding current treatments do not appear to be shared by the treating physicians highlighting a need for greater patient–doctor communication.
Classical treatment options for atopic dermatitis
Non-pharmacological management
Identification and avoidance of an individual’s trigger factors allows longer periods of remission or total clearance of symptoms. Numerous non-specific physical, chemical and biological factors can irritate the skin and elicit flares in patients with atopic dermatitis. Meanwhile, specific allergens can also promote skin lesions in sensitised patients18.
Figure 3: A selection of common non-specific (left) and specific (right) triggers seen in patients with atopic dermatitis (adapted from Wollenberg et al.18).
Environmental factors such as tobacco smoke and traffic exhaust have been shown to increase the risk of children developing atopic dermatitis and avoidance of them in young children has been introduced into the German S3 guidelines for the prevention of atopic diseases19.
Various strategies to avoid allergens have been proposed although with varying levels of evidence. While smooth clothing and avoidance of irritating fabrics and fibres are essential to avoid primary skin irritation other options include18:
- Mite-proof bedding and pyjamas
- Pollen avoidance
- Avoidance of relevant contact allergens e.g. fragrances, preservatives, emulsifiers
- Primary prevention of food allergy-associated adverse events with exclusive breast milk feeding until 4 months of age
- Observe a therapeutic diet eliminating those foods that elicited clinical early or late reactions upon controlled oral provocation tests
Emollients and skin care
Management of dry skin is critical to help maintain barrier function. The effective use of bathing and emollients are frequently recommended as an essential component of atopic dermatitis therapy18.
Cleansing and bathing
Cleansing the skin is essential to carefully remove dead skin and eliminate bacterial contaminants. However, it must be performed gently, and quickly to avoid irritation and epidermal dehydration. The use of bath oils, non-soap-based and acidic-cleansers as well as the addition of antiseptics have all been suggested to help manage atopic dermatitis18,20.
Emollients
Emollients represent the mainstay of atopic dermatitis management and typically contain a humectant to promote stratum corneum hydration, and an occludent to reduce evaporation.
It has been demonstrated that long-term emollient therapy improves xerosis associated with atopic dermatitis and the daily use of emollients from birth may significantly reduce the incidence of atopic dermatitis in a high-risk population. However, long-term follow-up data is required to confirm this18. Importantly, the use of emollients has been shown to be steroid sparing, reducing the side effect risks commonly associated with their prolonged use. A short-term steroid sparing effect has been observed in children and adults with mild-to-moderate atopic dermatitis following emollient use21,22,23, while long-term maintenance of stable disease has been achievable through regular use of emollients following induction of remission with TCS24,25.
Some caution should be exercised over the use of certain emollients in young children as some ingredients may cause irritation or under-desirable side effects. Urea can be used as a humectant but has been shown to cause irritation and kidney dysfunction in infants and should be avoided, while in toddlers, a lower concentration should be used than in adults. Meanwhile propylene glycol can irritate the skin of children aged below 2 years and so should not be used18. Concerns have also been raised about emollients containing intact proteins. While the use of colloidal oat meal has been shown to improve clinical outcomes in atopic dermatitis26, there are fears that it may increase the risk of skin sensitisation and allergy in at risk patients and so should be avoided in children below two years of age18.
Finally, it is worth considering that the sole use of emollients without sufficient topical anti-inflammatory therapy increases the risk of disseminated bacterial and viral infection – a risk that is already elevated in those with atopic dermatitis27.
Phototherapy
Patients with atopic dermatitis frequently observe an improvement in their symptoms during summer due to the increased sun exposure. During the course of the summer holidays, 74% of patients with mild-to-moderate atopic dermatitis saw complete resolution in one study. Interestingly, more patients saw complete resolution on seaside holidays (91%) than mountain holidays (11%)28. While UV exposure does not completely explain this difference, it supports the positive effects of UV radiation on atopic dermatitis18.
Table 2: Phototherapy options (adapted from Schäfer et al.18). UVA, ultraviolet A; UVB, ultraviolet B; NB-UVB, narrowband UVB; BB-UVB, broadband UVB; UVAB, combination of UVA and UVB; PUVA, psoralen plus UVA; UV, ultraviolet.
In Europe, NB-UVB has been used for patients with chronic, moderate forms of atopic dermatitis and is preferred to BB-UVB, while more severe cases have been treated with UVA118. Interestingly, a small number of patients with atopic dermatitis do not tolerate NB-UVB but respond well to BB-UVB29.
Mechanism of action
Ultraviolet radiation has been observed to produce a number of biological effects within the skin that may explain its ability to treat atopic dermatitis.
Figure 4: Proposed mechanisms of action for phototherapy in atopic dermatitis (adapted from Schäfer et al.18).
Phototherapy and pruritus
Use of narrowband UVB and UVA1 has been shown to be the most effective forms of UV therapy in multiple RCTs, including in the reduction of itch intensity. There is no anti-itch specific data for UV therapy though18.
Topical anti-inflammatory treatment
Topical anti-inflammatory treatments currently consist of topical corticosteroids (TCS) and topical calcineurin inhibitors (TCI). Their effective use requires18:
- Sufficient strength/potency
- Sufficient dosage
- Correct application
Traditionally, these conventional therapies were used reactively, applied to lesional skin and then stopped or tapered once visible lesions had cleared. However, a shift towards proactive treatment has been recommended where, once lesions have cleared, twice weekly application of the topical anti-inflammatory is continued alongside liberal use of emollients. Good results have been observed with topical corticosteroids and TCI, although topical corticosteroids are typically prescribed for short periods and no data beyond 20 weeks is available indicating an ongoing need for more research and additional therapeutic options to complement conventional therapy18.
Topical corticosteroids
A variety of topical corticosteroids (TCS) are available for patients with atopic dermatitis with the appropriate treatment based on patient-related factors, such as severity, patient age, the area of the body is to be applied to, and the physiochemical properties of the drug18,30. Local adverse events can be a concern with the use of TCS, but systemic effects are typically restricted to prolonged use and/or the use of high potency options30.
Potency
Many divide the available TCS into four classes not taking into account the type of formulation: mild, moderate, strong and very strong preparations2. However, the World Health Organization divides TCS into seven classes/groups based on potency, concentration and vehicle, meaning the same drug can be placed into different classes. These seven classes are then placed into four groups with group I being ultrahigh potency and group IV low potency30.
Table 3: Classification of topical corticosteroids by potency (adapted from Thomsen2).
When choosing a TCS18:
- Very strong (super potent) topical corticosteroids are not recommended for the management of atopic dermatitis
- Strong and very strong topical corticosteroids are more likely to cause adrenal function depression than mild and moderate topical corticosteroids. However, they will more rapidly restore skin barrier function and systemic effects will decrease more quickly
- Treatment of the face and eyelid region should be restricted to mild topical corticosteroids
- Children should be treated with less potent topical corticosteroids than adults
Mechanism of action
Corticosteroids have anti-proliferative, immunosuppressive, anti-inflammatory and vasoconstrictor actions. These effects can be divided into ‘genomic’ and ‘non-genomic’ mechanisms30.
The genomic mechanism of action involves corticosteroid binding to the nuclear glucocorticoid receptor resulting in changes to protein transcription. This process has been shown to induce transcription of anti-inflammatory proteins and have a broad range of effects on the cells of the immune system30.
Table 4: Corticosteroid effects on immune cells (adapted from Kwatra & Mukhopadhyay30).
While significant progress has been made in our understanding of the genomic effects topical corticosteroids has, less is known about the non-genomic mechanisms of action. However, it has been proposed that corticosteroid interactions at the plasma- and mitochondrial membranes could alter their physicochemical properties and activities of membrane-associated proteins30.
Topical calcineurin inhibitors
Two topical calcineurin inhibitors have been approved for the management of atopic dermatitis; tacrolimus ointment for moderate-to-severe disease and pimecrolimus cream for mild or moderate disease. While tacrolimus is comparable to a moderate to strong topical corticosteroids, pimecrolimus has a similar potency to a mild topical corticosteroid2.
Importantly, topical calcineurin inhibitors are not associated with thinning of the skin as seen with topical corticosteroids allowing them to be used for longer periods of time.
Long-term safety data has been published for each topical calcineurin inhibitor. A 4-year follow-up of patients using continuous or intermittent tacrolimus revealed a safety profile that was consistent with shorter periods of use indicating that long-term use gave no additional reason for concern31. Meanwhile, a 5-year follow-up of patients receiving topical corticosteroids with pimecrolimus or topical corticosteroid alone showed a similar profile and frequency of adverse events between the two groups while the addition of pimecrolimus was found to be steroid sparing13.
Both tacrolimus and pimecrolimus have also been shown to significantly reduce itch, with a 36% reduction compared to vehicle18. Importantly, topical calcineurin inhibitors are effective within 48 hours of application and show continued anti-pruritic effects with ongoing use. However, they are associated with burning/stinging upon application20.
However, some concerns have been raised with a commentary article highlighting that patients receiving pimecrolimus saw a higher incidence of bronchitis, infected eczema, impetigo and nasopharyngitis32.
Mechanism of action
Calcineurin inhibitors are effective immunosuppressants; oral tacrolimus is a widely used therapy for the prevention of transplant rejection. The topical calcineurin inhibitors function by suppressing the synthesis of pro-inflammatory cytokines in the skin. Both pimecrolimus and tacrolimus block the synthesis and release of IL-2, IL-3, IL-4, IL-5, interferon γ (IFNγ), and tumour necrosis factor α (TNFα) from T cells and Mast cells. However, tacrolimus also inhibits eosinophil and basophil activity, while blocking Langerhans cell function and promoting their apoptosis33.
Figure 5: Mechanism of action of topical calcineurin inhibitors (adapted from Breuer et al.34). IFN, interferon; IL, interleukin; NF-AT, nuclear factor of activated T cells; NF-ATc, NF-AT cytoplasm; NF-ATn, NF-AT nucleus; TCR, T cell receptor; TNF, tumour necrosis factor.
Pruritus
While anti-inflammatory rather than anti-pruritic, topical corticosteroids have been shown to induce a rapid itch reduction in atopic dermatitis18. A meta-analysis of six RCTs found a 34% reduction in itch versus vehicle35. However, a wide variety of topical corticosteroids with differing potencies and efficacies are available. Data suggests that moderate and high potency options are more effective than low potency topical steroids, but moderate and high potency options are similar20. While moderate use of topical corticosteroids should not have any systemic or local adverse events, patient concerns are common and should be addressed. By considering several factors such as potency, patient age and body area affected treatment can be optimised18.
Topical antihistamines
Limited data is available on the use of topical antihistamine formulations. A meta-analysis of four RCTs comparing topical antihistamines to vehicle (three evaluated doxepin 5% cream, one evaluated cromoglycate 4% lotion) identified a significant pooled reduction in pruritus of 27%35. However, topical doxepin is associated with an increased risk of contact allergy and so is not licensed or used in Europe18.
Targeted treatment options for atopic dermatitis
Non-steroidal topical anti-inflammatory treatment
The limited range of topical treatments for atopic dermatitis has led to great interest in novel targeted non-steroidal anti-inflammatory therapies based on a growing understanding of AD pathophysiology. Recently, phosphodiesterase-4 (PDE4) has become a desirable target for therapy due to its role in the inflammatory cascade of atopic dermatitis36.
Phosphodiesterase-4 (PDE4) inhibitors
Currently, a number of PDE4 inhibitors are under investigation for the management of atopic dermatitis due to their ability to reduce the production of proinflammatory cytokines37.
PDE4 inhibitors utilise a unique mechanism, differing from that of TCSs and TCIs, that affects a wide range of cytokines in atopic dermatitis36,38
The first to reach the market was crisaborole, a small lipophilic, boron-based molecule added to a topical vehicle that has been approved by the FDA for the treatment of mild-to-moderate atopic dermatitis in patients two years and older in the USA18. Crisaborole was also approved for use by the European medicines Agency (EMA) for the treatment of mild-to-moderate atopic dermatitis in adults and paediatric patients in 2020. However, in early 2022, marketing authorisastion was revoked and crisaborole therapy was not launched in the region39. Despite this, crisaborole maintains FDA approval and is widely used in the United States18.
The clinical trials that led to crisaborole’s FDA approval included AD-301 and AD-302, two phase 3, vehicle-controlled, double blind studies40. These trials enrolled 1527 patients, aged two years and over, with mild-to-moderate atopic dermatitis. The studies compared the study drug with a vehicle control twice daily for 28 days with a primary endpoint of clear or almost clear and a 2-grade or more improvement on the Investigator’s Static Global Assessment40. A post-hoc analysis pooling data from these two phase III trials saw significantly more patients report early improvement in pruritus with crisaborole than with vehicle (56.6% vs. 39.5%; p<0.001)41.
Crisaborole is the first topical PDE-4 inhibitor indicated for the treatment of mild-to-moderate AD. It is a safe and efficacious second-line option for the treatment of mild-to-moderate AD in patients 2 years of age and older42.
Other PDE4 inhibitors are also under development for atopic dermatitis, including topical therapies such as roflumilast, apremilast, GW842470X, E6005, AN2898 and OPA-15406 and the oral therapy roflumilast (all of which have currently completed Phase II studies and some Phase III trials)38.
More on current recommendations for the use of PDE4 inhibitors in the treatment of atopic dermatitis
Phosphodiesterase 4 regulates inflammatory cytokine production through the degradation of cyclic adenosine monophosphate (cAMP). In patients with atopic dermatitis, increased PDE4 activity leads to reduced cAMP levels and increased inflammation43. Selective inhibition of PDE4 prevents cAMP degrading to AMP. Increased levels of cAMP consequently suppress NF-kB which leads to the suppression of IFN-y and reduced inflammation36.
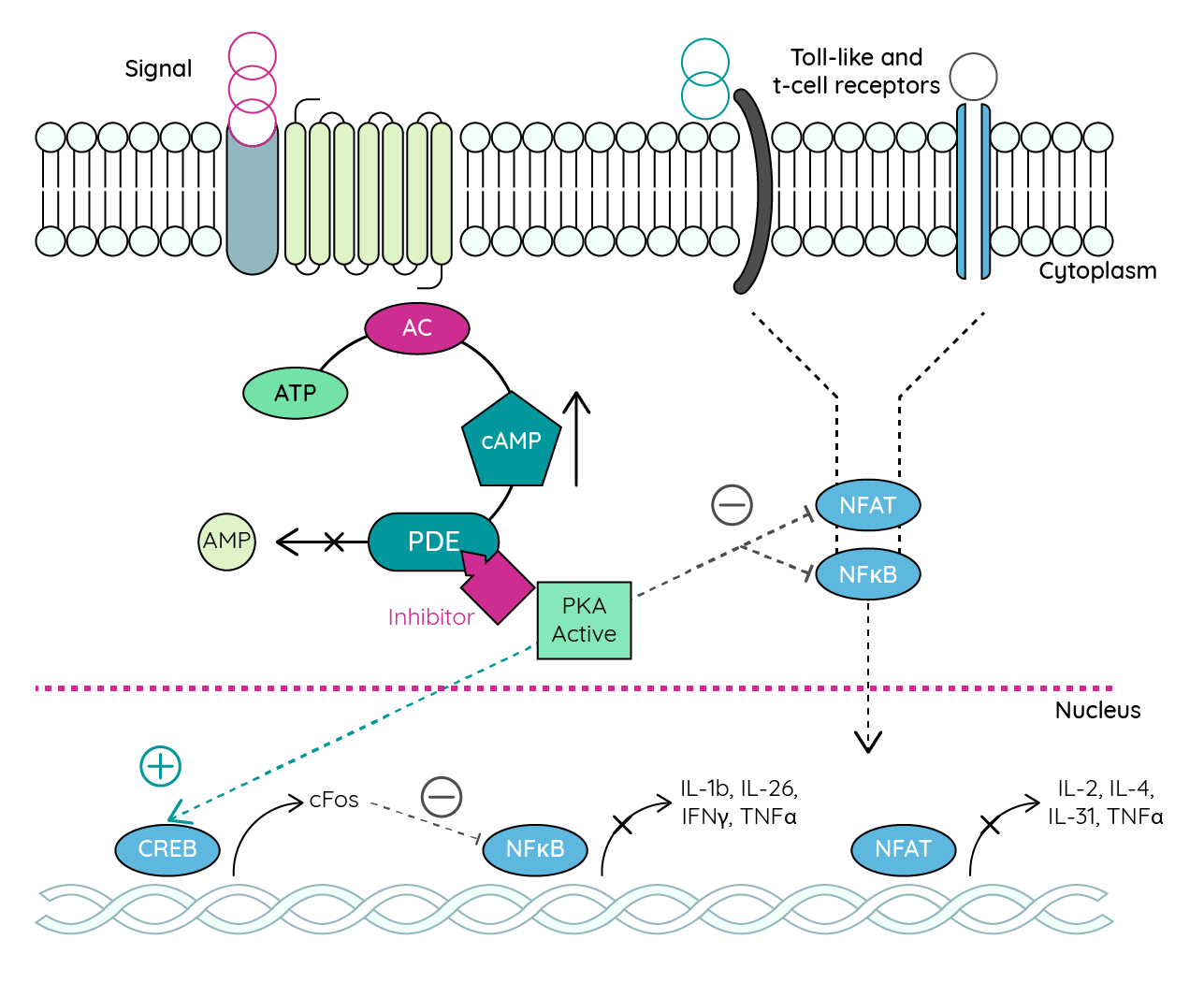
Figure 6: PDE4 inhibitor-mediated downregulation of proinflammatory cytokines (adapted from Ahluwalia et al.44).
AC adenylyl cyclase; CREB cAMP response element-binding protein; IL interleukin; NFAT nuclear factor of activated T-cells; NFjB nuclear factor kappa-light-chain-enhancer of activated B cells; PKA protein kinase A.
Discover more about the mechanism of action of PDE4 inhibitors with Professor Mike Cork
Systemic therapy
A variety of systemic treatments have been used for the treatment of severe atopic dermatitis, many of which are off label. Below we discuss the treatments licensed for this indication.
Oral glucocorticoids
Many European countries continue to use oral glucocorticoids for the management of atopic dermatitis18. However, systemic corticosteroids are not anti-pruritic and their prolonged use in atopic dermatitis is not recommended due to their safety profile20.
They should only be used short-term, preferably in combination with topical corticosteroids, to manage acute flares of severe, widespread disease. Given the risk of adverse effects, oral glucocorticoids should be tapered and replaced with an alternative immunosuppressant2. The use of systemic steroids in children should be undertaken with even greater caution than that with adults18.
The mechanism of action for oral glucocorticoids is consistent with that for topical corticosteroids, but experienced at a broader, systemic level.
Ciclosporin A
Ciclosporin A is an oral calcineurin inhibitor that promotes immunosuppression. In many European countries, it is a first-line treatment for adult patients with chronic severe atopic dermatitis who require systemic treatment.
While effective, treatment should not exceed two years and patients should be monitored for severe adverse effects. Common adverse effects include nephrotoxicity and hypertension with renal effects more likely if the daily dose exceeds 5 mg/kg, serum creatinine levels are elevated, or the patient is elderly18.
Ciclosporin A has a similar mechanism of action to the topical calcineurin inhibitors except it interacts with cyclophilin rather than macrophilin-1233.
Interleukin inhibition
Anti-IL-4/IL-13
Interleukin-4 (IL-4) and interleukin-13 (IL-13) are key Th2 cytokines that drive peripheral inflammation and pruritis in atopic dermatitis45. Inhibition of IL-4 and IL-13 can reduce itch and swelling by 50% in patients with moderate-to-severe atopic dermatitis46.
Monoclonal antibodies, dupilumab, approved in 2017 and tralokinumab, approved in 2021, are the latest biologic options for treating moderate-to-severe atopic dermatitis. Dupilumab exerts its effects by blocking IL-4 and IL-13, while tralokinumab selectively blocks IL-13.
A series of randomised controlled trials have confirmed the efficacy of dupilumab with a recent study confirming sustained activity over 1 year of continued treatment47,48,49. From the available data, approximately one-third of treated patients become clear or almost clear of atopic dermatitis based on the Investigator Global Assessment (IGA) and up to 70% of patients achieve an Eczema Area and Severity Index (EASI) 75 or higher improvement in their skin. Dupilumab also has an impressive safety profile with conjunctivitis the only adverse event observed more frequently than placebo18.
Similarly, the safety and efficacy of tralokinumab was investigated in the phase III ECZTRA 1 and ECZTRA 2 studies. Results showed that at 16 weeks of treatment, IGA and EASI 75 scores were significantly improved in treatment vs placebo, and this response was maintained at 52 weeks post-treatment. The safety profile of tralokinumab was comparable with placebo. Investigators concluded that tralokinumab significantly improves pruritis, sleep-disturbance, health related quality of life and severity of skin lesions50.
Lebrikizumab is another selective IL-13 inhibitor and is still under investigation in Phase III trials for moderate-to-severe atopic dermatitis51.
Mechanism of action
Interleukin-4 and IL-13 are critical components of the Th2-driven immune response observed in atopic dermatitis45. IL-13 plays a pivotal role in the production and maintenance of the inflammatory response and in the dysfunction of the epidermal barrier. Overexpression of IL-13 triggers downregulation in components necessary for epithelial integrity such as proteins and lipids as well as a loss of antimicrobial peptides. The resulting tissue inflammation causes thickening and remodelling of the skin, which when combined with the loss of antimicrobial peptides, increases the susceptibility to infections52. IL-13 has also been linked to pruritis through its sensitisation of pruritic neurons. Interestingly, biopsies of skin lesions have demonstrated overexpression of IL-13 and unremarkable amounts of IL-4, indicating that drugs that specifically target IL-13, such as tralokinumab and lebrikizumab, may be a more effective treatment52.
Future treatments
Several novel targeted, phenotype-specific therapies are on the horizon that may start an era of personalised therapy in atopic dermatitis43. This is of particular interest given the theory that early aggressive treatment of atopic dermatitis may help prevent the development of chronic disease and the atopic march53,54.
Anti-IL-5
IL-5 may be a target for future therapies for atopic dermatitis. The Th2 cytokine IL-5, acts within inflamed tissue to attract eosinophils and has been identified as playing a key role in eosinophilic asthma. Mepolizumab is an anti-IL-5 monoclonal antibody that has been approved for the treatment of severe eosinophilic asthma. While initial studies in patients with atopic dermatitis were underwhelming55, studies are ongoing looking at the benefits of anti-IL-5 therapy in patients with an increased eosinophil count51.
Figure 7: Action of IL-5 on eosinophil and inhibition via mepolizumab. Adapted from56.
Anti-IL-31
IL-31 is believed to play a role in the development of atopic dermatitis and pruritus and may be a target for therapy in the future. T helper 2 cells are the predominant producers of IL-31, which has been linked to intense pruritus in atopic dermatitis. The IL-31 receptor A is expressed on keratinocytes and a subset of dorsal root ganglions where the binding of IL-31 induces itch through the stimulation of sensory neurons.
An anti-IL-31 receptor A monoclonal antibody, nemolizumab, binds the receptor preventing IL-31 signalling and induction of pruritus. A phase II study of an anti-IL31 receptor A antibody, currently under development, showed significant reductions in pruritus in patients with moderate-to-severe atopic dermatitis57.
Figure 8: Inhibition of IL-31-mediated activation of gene transcription (adapted from Paller et al.51).
Figure 9: Dupilumab inhibition of IL-4 and IL-13 signalling (adapted from Barranco et al.58). IL, interleukin; JAK, Janus kinase; STAT, signal transducer and activator of transcription; Tyk, tyrosine kinase.
Oral phosphodiesterase-4 inhibitors (PDE-4)
Apremilast, an oral, small molecule PDE-4 inhibitor has been approved for the treatment of psoriatic arthritis and moderate-to-severe plaque psoriasis. A pilot study in 2012, investigated the effects of apremilast in patients with moderate-to-severe atopic dermatitis, showed moderate improvement of skin lesions, pruritus and quality of life59. However, the drug development programme of apremilast for atopic dermatitis is no longer under way18.
Janus kinases (JAK) inhibitors
Janus kinases (JAK) are a family of intracellular tyrosine kinases that signal via the Signal Transducer and Activator of Transcription (STAT) pathway to elicit the physiological effects of a variety of proinflammatory cytokines including IL-4, IL-5, IL-13 and IL-31. There are a number of JAK inhibitors which have been approved by the Food and Drug Administration (FDA) and European Medicines Agency (EMA) for use in moderate-to-severe atopic dermatitis, including: abrocitinib, upadacitinib and baricitinib60. Ruxolitinib is a topical JAK inhibitor which has demonstrated promising results, it has been approved for use by the FDA in the United States, however, is still undergoing assessment and validation from the EMA61.
Systemic JAK inhibitors
Abrocitinib
The approval of abrocitinib was based on results of five clinical trials from a large-scale program recruiting over 1,600 patients. The largest of these trials were the JADE clinical trials where abrocitinib was compared to placebo and combination with dupilumab for its safety and efficacy62. Results from the study showed a 75% improvement in Eczema Area and Severity Index response, and/or at least 4-point improvement in Peak Pruritus Numerical Rating Scale response in abrocitinib treated patients. Serious adverse events were reported for fewer than 3% of participants62. The recommended doses are 100 mg and 200 mg, with the 200-mg dose recommended for patients who are not responding to the 100-mg dose. Overall, abrocitinib has demonstrated a consistent safety profile and profound improvements in skin clearance, extent of disease, and severity, as well as rapid improvement in itch after only two weeks62–64.
Upadacitinib
Upadacitinib was approved for 15 mg and 30 mg doses, supported by efficacy and safety data from a large Phase III clinical trial program, recruiting 2,500 atopic dermatitis patients and evaluated across three studies65. Upadacitinib demonstrated significant improvement in itch as early as week one, as well as significant improvements in skin clearance (EASI 75 and vIGA-AD 0/1) at 16 weeks when compared to placebo65.
Baricitinib
Approval of baricitinib for atopic dermatitis was based on three phase III randomised, double-blind, placebo-controlled studies (BREEZE-AD1, BREEZE-AD2 and BREEZE-AD3)66. Barcitinib 2 mg or 4 mg was prescribed alone or in combination with topical treatments in adults with moderate-to-severe atopic-dermatitis, for whom topical treatments were ineffective or poorly tolerated. In all three studies, baricitinib was shown to be more effective than placebo in achieving skin clearance at 16 weeks. Barcitinib demonstrated sustained long-term efficacy with minimal adverse effects66.
Topical JAK inhibitors
Ruxolitinib
Ruxolitinib is the first topical JAK inhibitor cream to be approved by the FDA for the short-term treatment of atopic dermatitis61. Approval was based on data from the TRuE-AD clinical trial program, consisting of 2 randomised, double-blind, vehicle-controlled phase III studies evaluating the safety and efficacy of the cream in more than 1200 adolescents and adults with mild-to-moderate atopic dermatitis. 53.8% of patients in the treatment group achieved IGA treatment success at 8 weeks when compared to 7.6 % of placebo. Safety of ruxolitinib was not significantly different to that of placebo61.
Delgocitinib
Delgocitinib ointment was recently approved in Japan for paediatric and adult AD and is still undergoing phase III clinical trials before being approved in Europe and the US. In Japanese studies, delgocitinib ointment was effective and well tolerated in adult and paediatric patients with moderate to severe AD for up to 28 weeks67.
Professor Seemal Desai explains the importance of Janus kinase proteins in the context of a need to explore and find more targeted pathways for effective treatment of atopic dermatitis.
The safety and efficacy of JAK inhibitors have been thoroughly explored in Phase II and III trials. Professor Desai takes us through some of these investigational molecules which have great potential for the treatment of atopic dermatitis.
Oral antihistamines
Chronic pruritus, as seen in atopic dermatitis, is induced by the nonhistaminergic pathway20. Despite being used for decades, few randomised controlled trials (RCTs) have evaluated their efficacy in atopic dermatitis and those that have typically show a weak or no effect on pruritus. While the sedative effects of first-generation antihistamines may allow improved sleep, the evidence-base for an anti-pruritic effect of first- and second-generation antihistamines remains limited18.
Treating mild-to-moderate atopic dermatitis
Early treatment of atopic dermatitis is key to optimal treatment outcome in treating skin disease and may also delay or prevent the atopic march. There is limited data available regarding the specific clinical and economic burden of mild-to-moderate atopic dermatitis, however a possible link has been proposed between disease burden and disease severity68. Therefore, optimal management of mild-to-moderate presentation of this condition is essential.
Classical and targeted treatments each have a role to play in the treatment of mild-to-moderate manifestation of atopic dermatitis. Research continues to establish the most effective treatments that might be or become available to reduce symptoms and allow clinicians and patients to establish effective management.
A systematic review and meta-analysis of the application of topical phosphodiesterase 4 (PDE4) inhibitors in mild-to-moderate atopic dermatitis was conducted in 2019. The results indicated favourable efficacy and safety of PDE4 inhibitors, especially crisaborole and AN289869.
PDE4 inhibitor research continues into their use, particularly in children. Professor Michael Cork explains more, with reference to new technology to evaluate the effect that both PDE4 inhibitors and topical corticosteroids are having on the skin.
In 2019, a clinical review report was published to provide an overview and critical appraisal of the evidence available (both published and unpublished) for assessing the efficacy and harms of crisaborole (2%) ointment to the topical pharmacological therapies available. This review compared the different therapies in patients with mild-to-moderate AD but due to the limitation in the analyses no definitive conclusion was reached on the efficacy and safety of crisaborole to other topical therapies70.
Treating moderate-to-severe atopic dermatitis
There is a clear need for the continued development of targeted therapies, including biologics, which can address specific cytokine pathways implicated in atopic dermatitis. Biologics include those targeting Th2, Th22, TH17/IL-23 and IgE.
Guidelines recommend the anti-inflammatory biologic dupilumab, which targets IL-4 and IL-13, for the treatment of moderate-to-severe atopic dermatitis in adults and adolescents 12 years and older who are candidates for systemic therapy, and the treatment of severe atopic dermatitis in children 6 to 11 years old who are candidates for systemic therapy71. Licensed for use in the U.S. and Europe, dupilumab has been shown to hold significant efficacy in improving signs and symptoms of atopic dermatitis, with an acceptable safety profile46,47,49,72,73.
Tralokinumab, which selectively targets IL-13, a key driver of peripheral inflammation, has been demonstrated to be efficacious in the treatment of atopic dermatitis in large phase 3 studies, and has recently been approved by the FDA and EMA for use in adults eligible for systemic therapy50,52.
Oral JAK inhibitors have recently been approved for use in Europe and the United States, and when combined with topical agents, offer the most promising targeted treatment for moderate-to-severe AD, especially for people who have a suboptimal response to dupilumab60. Of note is abrocitinib, approved in early 2022 with outstanding efficacy and no serious safety concerns62. Short-term head-to-head studies of upadacitinib and abrocitinib vs dupilumab suggest a more rapid onset of efficacy and itch relief, and equivalent or better efficacy compared with dupilumab in EASI90, IGA0, or IGA1 end points65,74. However, as these treatments have only been recently approved, official guidelines have not yet been adapted to consider this emerging evidence.
Emerging topical and systemic therapies are being developed to primarily target the type 2 immune pathway. Topical treatments being investigated include ruxolitinib and delgocitinib (JAK inhibitors), tapinarof (an aryl hydrocarbon receptor agonist), and creams aimed at correcting microbial dysbiosis75. In addition to monoclonal antibodies targeting IL-4 and IL-13 (dupilumab and tralokinumab) other biologics targeting IL-13, IL-31, IL-33, OX40, and thymic stromal lymphopoietin are currently being tested in clinical trials75.
Professor Thomas Bieber provides an overview of new treatments, including biologics and small molecules, which are currently in the pipeline. Many have reached phase 3 trials. Biologics targeting interleukins such as IL-31 offer hope for the alleviation of chronic pruritis, and others focus on the inflammatory processes at play in atopic dermatitis. JAK kinase inhibitors are discussed as part of upcoming systemic and topical treatment.
A recent systematic review and meta-analysis provided evidence that JAK inhibitors have favourable efficacy in the treatment of AD with tolerable safety issues. However, it is thought that long-term follow-up of adverse events and cost-effectiveness could help make decisions in using JAK inhibitors for treating AD76.
Watch Professor Bieber expand on the potential they provide.
References
- Megna M, Napolitano M, Patruno C, Villani A, Balato A, Monfrecola G, et al. Systemic Treatment of Adult Atopic Dermatitis: A Review. Dermatol Ther (Heidelb). 2017;7(1):1–23.
- Thomsen SF. Atopic Dermatitis: Natural History, Diagnosis, and Treatment. ISRN Allergy. 2014;2014:1–7.
- Balieva FN, Finlay AY, Kupfer J, Tomas Aragones L, Lien L, Gieler U, et al. The role of therapy in impairing quality of life in dermatological patients: A multinational study. Acta Derm Venereol. 2018;98(6):563–569.
- George SMC, Makrygeorgou A. 8th Georg Rajka International Symposium on Atopic Dermatitis: meeting report. Br J Dermatol. 2015;172(4):916–925.
- Zane LT, Chanda S, Jarnagin K, Nelson DB, Spelman L, Gold LFS. Crisaborole and its potential role in treating atopic dermatitis: Overview of early clinical studies. Immunotherapy. 2016;8(8):853–866.
- Aubert-Wastiaux H, Moret L, Le Rhun A, Fontenoy AM, Nguyen JM, Leux C, et al. Topical corticosteroid phobia in atopic dermatitis: A study of its nature, origins and frequency. Br J Dermatol. 2011;165(4):808–814.
- Lee JY, Her Y, Kim CW, Kim SS. Topical corticosteroid phobia among parents of children with atopic eczema in Korea. Ann Dermatol. 2015;27(5):499–506.
- Aubert-Wastiaux H, Anthoine E, Moret L, Segard C, Bernier C, Barbarot S, et al. Are caregivers corticophobics? Br J Dermatol. 2014;170(6):e14–e15.
- Raffin D, Giraudeau B, Samimi M, Machet L, Pourrat X, Maruani A. Corticosteroid phobia among pharmacists regarding atopic dermatitis in children: A national French survey. Acta Derm Venereol. 2016;96(2):177–180.
- Moret L, Anthoine E, Aubert-Wastiaux H, Le Rhun A, Leux C, Mazereeuw-Hautier J, et al. TOPICOP©: A New Scale Evaluating Topical Corticosteroid Phobia among Atopic Dermatitis Outpatients and Their Parents. PLoS One. 2013;8(10). doi:10.1371/journal.pone.0076493.
- Stalder JF, Aubert H, Anthoine E, Futamura M, Marcoux D, Morren MA, et al. Topical corticosteroid phobia in atopic dermatitis: International feasibility study of the TOPICOP score. Allergy Eur J Allergy Clin Immunol. 2017;72(11):1713–1719.
- Werfel T. Topical use of pimecrolimus in atopic dermatitis. JDDG - J Ger Soc Dermatology. 2009;7(9):739–742.
- Sigurgeirsson B, Boznanski A, Todd G, Vertruyen A, Schuttelaar MLA, Zhu X, et al. Safety and efficacy of pimecrolimus in atopic dermatitis: A 5-year randomized trial. Pediatrics. 2015;135(4):597–606.
- McAlister RO, Tofte SJ, Doyle JJ, Jackson A, Hanifin JM. Patient and Physician Perspectives Vary on Atopic Dermatitis - PubMed. Cutis. 2002;69(6):461–466.
- Paller AS, Mcalister RO, Doyle JJ, Jackson A. Perceptions of physicians and pediatric patients about atopic dermatitis, its impact, and its treatment. Clin Pediatr (Phila). 2002;41(5):323–332.
- Murota H, Takeuchi S, Sugaya M, Tanioka M, Onozuka D, Hagihara A, et al. Characterization of socioeconomic status of Japanese patients with atopic dermatitis showing poor medical adherence and reasons for drug discontinuation. J Dermatol Sci. 2015;79(3):279–287.
- Schmitt J, Csötönyi F, Bauer A, Meurer M. Determinants of treatment goals and satisfaction of patients with atopic eczema. JDDG - J Ger Soc Dermatology. 2008;6(6):458–465.
- Wollenberg A, Barbarot S, Bieber T, Christen-Zaech S, Deleuran M, Fink-Wagner A, et al. Consensus-based European guidelines for treatment of atopic eczema (atopic dermatitis) in adults and children: part I. J Eur Acad Dermatology Venereol. 2018;32(5):657–682.
- Schäfer T, Bauer C-P, Beyer K, Bufe A, Friedrichs F, Gieler U, et al. S3-Guideline on allergy prevention: 2014 update. Allergo J Int. 2014;23(6):186–199.
- Mollanazar NK, Smith PK, Yosipovitch G. Mediators of Chronic Pruritus in Atopic Dermatitis: Getting the Itch Out? Clin Rev Allergy Immunol. 2016;51(3):263–292.
- Grimalt R, Mengeaud V, Cambazard F. The steroid-sparing effect of an emollient therapy in infants with atopic dermatitis: A randomized controlled study. Dermatology. 2006;214(1):61–67.
- Eberlein B, Eicke C, Reinhardt HW, Ring J. Adjuvant treatment of atopic eczema: Assessment of an emollient containing N-palmitoylethanolamine (ATOPA study). J Eur Acad Dermatology Venereol. 2008;22(1):73–82.
- Szczepanowska J, Reich A, Szepietowski JC. Emollients improve treatment results with topical corticosteroids in childhood atopic dermatitis: A randomized comparative study. Pediatr Allergy Immunol. 2008;19(7):614–618.
- Mengeaud V, Phulpin C, Bacquey A, Boralevi F, Schmitt AM, Taieb A. An innovative oat-based sterile emollient cream in the maintenance therapy of childhood atopic dermatitis. Pediatr Dermatol. 2015;32(2):208–215.
- Åkerström U, Reitamo S, Langeland T, Berg M, Rustad L, Korhonen L, et al. Comparison of moisturizing creams for the prevention of atopic dermatitis relapse: A randomized double-blind controlled multicentre clinical trial. Acta Derm Venereol. 2015;95(5):587–592.
- Fowler JF, Nebus J, Wallo W, Eichenfield LF. Colloidal Oatmeal Formulations as Adjunct Treatments in Atopic Dermatitis. J Drugs Dermatol. 2012;11(7):804–807.
- Wollenberg A, Wetzel S, Burgdorf WHC, Haas J. Viral infections in atopic dermatitis: Pathogenic aspects and clinical management. J Allergy Clin Immunol. 2003;112(4):667–674.
- Patrizi A, Savoia F, Giacomini F, Tabanelli M, Gurioli C, Romagnoli A, et al. The effect of summer holidays and sun exposure on atopic dermatitis. G Ital di Dermatologia e Venereol. 2009;144(4):463–466.
- Pugashetti R, Lim HW, Koo J. Broadband UVB revisited: Is the narrowband UVB fad limiting our therapeutic options? J Dermatolog Treat. 2010;21(6):326–330.
- Kwatra G, Mukhopadhyay S. Topical corticosteroids: Pharmacology. In: A Treatise on Topical Corticosteroids in Dermatology: Use, Misuse and Abuse. 2017. Springer Singapore: 11–22.
- Reitamo S, Rustin M, Harper J, Kalimo K, Rubins A, Cambazard F, et al. A 4-year follow-up study of atopic dermatitis therapy with 0.1% tacrolimus ointment in children and adult patients. Br J Dermatol. 2008;159(4):942–951.
- Weidinger S, Baurecht H, Schmitt J. A 5-year randomized trial on the safety and efficacy of pimecrolimus in atopic dermatitis: a critical appraisal. Br J Dermatol. 2017;177(4):999–1003.
- Gutfreund K, Bienias W, Szewczyk A, Kaszuba A. Topical calcineurin inhibitors in dermatology. Part I: Properties, method and effectiveness of drug use. Postep Dermatologii i Alergol. 2013;30(3):165–169.
- Breuer K, Werfel T, Kapp A. Safety and efficacy of topical calcineurin inhibitors in the treatment of childhood atopic dermatitis. American Journal of Clinical Dermatology. 2005;6(2):65–77.
- Sher LG, Chang J, Patel IB, Balkrishnan R, Fleischer AB. Relieving the pruritus of atopic dermatitis: A meta-analysis. Acta Derm Venereol. 2012;92(5):455–461.
- Guttman-Yassky E, Hanifin JM, Boguniewicz M, Wollenberg A, Bissonnette R, Purohit V, et al. The role of phosphodiesterase 4 in the pathophysiology of atopic dermatitis and the perspective for its inhibition. Exp Dermatol. 2019;28(1):3–10.
- Fishbein AB, Silverberg JI, Wilson EJ, Ong PY. Update on Atopic Dermatitis: Diagnosis, Severity Assessment, and Treatment Selection. J Allergy Clin Immunol Pract. 2020;8(1):91–101.
- Munera-Campos M, Carrascosa JM. Innovation in Atopic Dermatitis: From Pathogenesis to Treatment. Actas Dermo-Sifiliograficas. 2020;111(3). doi:10.1016/j.ad.2019.11.002.
- European Medicines Agency (EMA). Staquis 20 mg/g ointment SmPC Available at: https://www.ema.europa.eu/en/documents/product-information/staquis-epar-product-information_en.pdf. Accessed 21 April 2021.
- Paller AS, Tom WL, Lebwohl MG, Blumenthal RL, Boguniewicz M, Call RS, et al. Efficacy and safety of crisaborole ointment, a novel, nonsteroidal phosphodiesterase 4 (PDE4) inhibitor for the topical treatment of atopic dermatitis (AD) in children and adults. J Am Acad Dermatol. 2016;75(3):494-503.e6.
- Yosipovitch G, Stein Gold LF, Lebwohl MG, Silverberg JI, Tallman AM, Zane LT. Early relief of pruritus in atopic dermatitis with crisaborole ointment, a non-steroidal, phosphodiesterase 4 inhibitor. Acta Derm Venereol. 2018;98(5):484–489.
- McDowell L, Olin B. Crisaborole: A Novel Nonsteroidal Topical Treatment for Atopic Dermatitis. Journal of Pharmacy Technology. 2019;35(4):172–178.
- Cabanillas B, Brehler AC, Novak N. Atopic dermatitis phenotypes and the need for personalized medicine. Current Opinion in Allergy and Clinical Immunology. 2017;17(4):309–315.
- Ahluwalia J, Udkoff J, Waldman A, Borok J, Eichenfield LF. Phosphodiesterase 4 Inhibitor Therapies for Atopic Dermatitis: Progress and Outlook. Drugs. 2017;77(13):1389–1397.
- Wong LS, Wu T, Lee CH. Inflammatory and noninflammatory itch: Implications in pathophysiology-directed treatments. Int J Mol Sci. 2017;18(7). doi:10.3390/ijms18071485.
- Beck LA, Thaçi D, Hamilton JD, Graham NM, Bieber T, Rocklin R, et al. Dupilumab Treatment in Adults with Moderate-to-Severe Atopic Dermatitis. N Engl J Med. 2014;371(2):130–139.
- Thaçi D, Simpson EL, Beck LA, Bieber T, Blauvelt A, Papp K, et al. Efficacy and safety of dupilumab in adults with moderate-to-severe atopic dermatitis inadequately controlled by topical treatments: A randomised, placebo-controlled, dose-ranging phase 2b trial. Lancet. 2016;387(10013):40–52.
- Simpson E, Bieber T, Guttman-Yassky E, Beck L, Blauvelt A, Cork M. Two Phase 3 Trials of Dupilumab versus Placebo in Atopic Dermatitis. N Engl J Med. 2016;375(24):2335–2348.
- Blauvelt A, de Bruin-Weller M, Gooderham M, Cather JC, Weisman J, Pariser D, et al. Long-term management of moderate-to-severe atopic dermatitis with dupilumab and concomitant topical corticosteroids (LIBERTY AD CHRONOS): a 1-year, randomised, double-blinded, placebo-controlled, phase 3 trial. Lancet. 2017;389(10086):2287–2303.
- Wollenberg A, Blauvelt A, Guttman-Yassky E, Worm M, Lynde C, Lacour JP, et al. Tralokinumab for moderate-to-severe atopic dermatitis: results from two 52-week, randomized, double-blind, multicentre, placebo-controlled phase III trials (ECZTRA 1 and ECZTRA 2). Br J Dermatol. 2021;184(3):437–449.
- Paller AS, Kabashima K, Bieber T. Therapeutic pipeline for atopic dermatitis: End of the drought? J Allergy Clin Immunol. 2017;140(3):633–643.
- Gon alves F, Freitas E dio, Torres T. Selective IL-13 inhibitors for the treatment of atopic dermatitis. Drugs Context. 2021;10. doi:10.7573/DIC.2021-1-7.
- Bieber T, Cork M, Reitamo S. Atopic dermatitis: A candidate for disease-modifying strategy. Allergy Eur J Allergy Clin Immunol. 2012;67(8):969–975.
- McPherson T. Current understanding in pathogenesis of atopic dermatitis. Indian J Dermatol. 2016;61(6):649–655.
- Oldhoff JM, Darsow U, Werfel T, Katzer K, Wulf A, Laifaoui J, et al. Anti-IL-5 recombinant humanized monoclonal antibody (Mepolizumab) for the treatment of atopic dermatitis. Allergy Eur J Allergy Clin Immunol. 2005;60(5):693–696.
- Pelaia C, Vatrella A, Busceti MT, Gallelli L, Terracciano R, Savino R, et al. Severe eosinophilic asthma: From the pathogenic role of interleukin-5 to the therapeutic action of mepolizumab. Drug Design, Development and Therapy. 2017;11:3137–3144.
- Ruzicka T, Hanifin JM, Furue M, Pulka G, Mlynarczyk I, Wollenberg A, et al. Anti–Interleukin-31 Receptor A Antibody for Atopic Dermatitis. N Engl J Med. 2017;376(9):826–835.
- Barranco P, Phillips-Angles E, Dominguez-Ortega J, Quirce S. Dupilumab in the management of moderate-to-severe asthma: The data so far. Ther Clin Risk Manag. 2017;13:1139–1149.
- Samrao A, Berry TM, Goreshi R, Simpson EL. A pilot study of an oral phosphodiesterase inhibitor (apremilast) for atopic dermatitis in adults. Arch Dermatol. 2012;148(8):890–897.
- Chovatiya R, Paller AS. JAK inhibitors in the treatment of atopic dermatitis. J Allergy Clin Immunol. 2021;148(4):927–940.
- Papp K, Szepietowski JC, Kircik L, Toth D, Eichenfield LF, Leung DYM, et al. Efficacy and safety of ruxolitinib cream for the treatment of atopic dermatitis: Results from 2 phase 3, randomized, double-blind studies. J Am Acad Dermatol. 2021;85(4):863–872.
- Alexis A, de Bruin-Weller M, Weidinger S, Soong W, Barbarot S, Ionita I, et al. Rapidity of Improvement in Signs/Symptoms of Moderate-to-Severe Atopic Dermatitis by Body Region with Abrocitinib in the Phase 3 JADE COMPARE Study. Dermatol Ther (Heidelb). 2022;12(3):771–785.
- Silverberg JI, Simpson EL, Thyssen JP, Gooderham M, Chan G, Feeney C, et al. Efficacy and Safety of Abrocitinib in Patients With Moderate-to-Severe Atopic Dermatitis: A Randomized Clinical Trial. JAMA dermatology. 2020;156(8):863–873.
- Simpson EL, Sinclair R, Forman S, Wollenberg A, Aschoff R, Cork M, et al. Efficacy and safety of abrocitinib in adults and adolescents with moderate-to-severe atopic dermatitis (JADE MONO-1): a multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet. 2020;396(10246):255–266.
- Guttman-Yassky E, Teixeira HD, Simpson EL, Papp KA, Pangan AL, Blauvelt A, et al. Once-daily upadacitinib versus placebo in adolescents and adults with moderate-to-severe atopic dermatitis (Measure Up 1 and Measure Up 2): results from two replicate double-blind, randomised controlled phase 3 trials. Lancet (London, England). 2021;397(10290):2151–2168.
- Silverberg JI, Simpson EL, Wollenberg A, Bissonnette R, Kabashima K, Delozier AM, et al. Long-term Efficacy of Baricitinib in Adults With Moderate to Severe Atopic Dermatitis Who Were Treatment Responders or Partial Responders: An Extension Study of 2 Randomized Clinical Trials. JAMA dermatology. 2021;157(6):691–699.
- Nakagawa H, Nemoto O, Igarashi A, Saeki H, Kaino H, Nagata T. Delgocitinib ointment, a topical Janus kinase inhibitor, in adult patients with moderate to severe atopic dermatitis: A phase 3, randomized, double-blind, vehicle-controlled study and an open-label, long-term extension study. J Am Acad Dermatol. 2020;82(4):823–831.
- Toron F, Neary MP, Smith TW, Gruben D, Romero W, Cha A, et al. Clinical and Economic Burden of Mild-to-Moderate Atopic Dermatitis in the UK: A Propensity-Score-Matched Case–Control Study. Dermatol Ther (Heidelb). 2021;11(3):907–928.
- Yang H, Wang J, Zhang X, Zhang Y, Qin ZL, Wang H, et al. Application of Topical Phosphodiesterase 4 Inhibitors in Mild to Moderate Atopic Dermatitis: A Systematic Review and Meta-analysis. JAMA Dermatology. 2019;155(5):585–593.
- CADTH. Summary of Indirect Comparisons - Clinical Review Report: Crisaborole Ointment, 2% (Eucrisa) - NCBI Bookshelf. 2019. https://www.ncbi.nlm.nih.gov/books/NBK542340/. Accessed 11 June 2021.
- Summary of Product Characteristics. Cosentyx 150 mg solution for injection in pre-filled pen. https://www.medicines.org.uk/emc/product/9380/smpc%0Ahttps://www.medicines.ie/medicines/cosentyx-150-mg-solution-for-injection-in-pre-filled-pen-31719/smpc. Accessed 23 April 2021.
- Simpson EL, Bieber T, Guttman-Yassky E, Beck LA, Blauvelt A, Cork MJ, et al. Two Phase 3 Trials of Dupilumab versus Placebo in Atopic Dermatitis. N Engl J Med. 2016;375(24):2335–2348.
- de Bruin-Weller M, Thaçi D, Smith CH, Reich K, Cork MJ, Radin A, et al. Dupilumab with concomitant topical corticosteroid treatment in adults with atopic dermatitis with an inadequate response or intolerance to ciclosporin A or when this treatment is medically inadvisable: a placebo-controlled, randomized phase III clinical t. Br J Dermatol. 2018;178(5):1083–1101.
- Bieber T, Simpson EL, Silverberg JI, Thaçi D, Paul C, Pink AE, et al. Abrocitinib versus Placebo or Dupilumab for Atopic Dermatitis. N Engl J Med. 2021;384(12):1101–1112.
- Puar N, Chovatiya R, Paller AS. New treatments in atopic dermatitis. Ann Allergy, Asthma Immunol. 2021;126(1):21–31.
- Tsai HR, Lu JW, Chen LY, Chen TL. Application of janus kinase inhibitors in atopic dermatitis: An updated systematic review and meta-analysis of clinical trials. Journal of Personalized Medicine. 2021;11(4). doi:10.3390/jpm11040279.
of interest
are looking at
saved
next event
This content has been developed by Medthority who previously received funding in order to help provide its healthcare professional members with access to the highest quality medical and scientific information, education and associated relevant content.